目的 从虎眼万年青中提取、分离水溶性多糖,初步研究其特征和抗肿瘤活性。 方法 采用热水提取,乙醇沉淀,Sevag法脱蛋白,DEAE-Sepharose Fast Flow离子交换柱色谱和Sephadex G-75凝胶过滤柱色谱分离纯化,得到虎眼万年青均一多糖OCAP-2-2。毛细管区带电泳法(CZE)分析单糖组成;采用高效凝胶渗透色谱法(HPGPC)测定多糖纯度和相对分子质量;动物移植性实体瘤的瘤重实验法研究对小鼠S180肉瘤的抑瘤作用,采用细胞体外培养技术,MTT法检测对人白血病细胞株K562细胞的增殖抑制作用。 结果 经过分离纯化得到的均一多糖组分OCAP-2-2主要由葡萄糖、木糖、甘露糖、半乳糖等4种单糖组成,其相对分子质量之比为2.16∶1.26:0.88∶1.00,平均相对分子质量9.84×104,总糖含量为92.3%,总糖醛酸含量为6.21%,蛋白质含量为3.68%;在0.1~100 μgmL-1内与荷瘤对照组相比,OCAP-2-2对小鼠S180肉瘤有显著的抑制活性,其中多糖浓度为100 μgmL-1时抑瘤率达(53.16±4.23)% ([i]P<[/i]0.001);OCAP-2-2对K562细胞有明显的增殖抑制作用([i]P<[/i]0.01),在多糖浓度为0.10 μgmL-1时,增殖抑制率最高为(39.83±7.31)% ([i]P<[/i]0.01)。 结论 OCAP-2-2具有很高的抗肿瘤活性,可以探索作为一种潜在的天然抗肿瘤药物。
各位前辈好!最近想买用来分离β-葡聚糖的凝胶填料柱,分子量范围大概在8x104~1.6x106之间,看网上和书上的凝胶填料范围大多数写的是分离的蛋白质分子量的,有点小纠结了,这个是通用的吗?以蛋白质的分子量来看选择纯化多糖的填料准吗?大家有木有合适的、用的好的凝胶填料厂家,或者说明书啥的哈,谢谢各位了!
[align=center][/align][font=宋体]一、原理[/font][font=宋体]1.[/font][font=宋体]多糖的分级与纯化[/font][font=宋体]多糖纯化的实质是将粗多糖中的杂质(包括蛋白质、色素、低聚糖、无机盐等非糖物质)除去而获得分子量和极性均一的多糖组分。比较常用的方法有:醇沉法、超滤法和柱层析法等。其中柱层析法应用比较普遍,多用于样品的精细分级,具有操作简单,重现性好的优点,但样品上样量小。用于多糖分离的柱层析主要有两类:一类是通过分子量大小进行分级的凝胶柱层析,如Sephadex G-100(200、50、25、10)、SepharoseCL-6B等系列;另一类是离子交换层析,是根据多糖所带电荷的性质不同选择相应的离子交换柱对多糖进行分级。如待分离的多糖带有负电荷,可选择阴离子型的DEAE-纤维素柱或DEAE-Sepharose柱进行分级,以获得均一的多糖组分。[/font][font=宋体]通过热水煮提方法提取的多糖,通常是混合物,且分子量不均一,其单糖组成、分子结构和聚合度往往不同,可通过柱层析的方法,对其进行分级以达纯化的目的。本实验中,采用[/font]DEAE-[font=宋体]纤维素柱层析的方法对中草药中水溶性粗多糖进行分级纯化,分离纯化出各级分多糖,[/font][font=宋体]为后续研究多糖的结构特征和生物活性奠定基础。[/font][font=宋体]2.[/font][font=宋体]苯酚—硫酸法[/font][font=宋体]游离的寡糖、多糖中的己糖或糖醛酸在浓硫酸的作用下,脱水生成糠醛,再与苯酚作用起显色反应,在一定范围内其颜色深浅与糖的含量成正比,吸收值与糖含量呈线性关系。且己糖在[/font]490 nm[font=宋体]处(戊糖及糖醛酸在[/font]480 nm[font=宋体])有最大吸收,故用比色法测定多糖含量。该法简单,快速,灵敏,且颜色持久,同一台设备同一光源仅需制作一条标准曲线。[/font][font=宋体] [/font][font=宋体]二、步骤[/font]1.[font=宋体]中草药水溶粗多糖的提取流程[/font][font=宋体]中草药粉末[/font][font=Symbol][/font][font=宋体]蒸馏水浸泡过夜[/font][font=Symbol][/font][font=宋体]超声[/font]/[font=宋体]微波[/font][font=Symbol][/font][font=宋体]水浴浸提[/font][font=Symbol][/font][font=宋体]过滤去除药渣[/font][font=Symbol][/font][font=宋体]定容[/font][font=Symbol][/font][font=宋体]多糖含量测定[/font][font=宋体]加等体积[/font]95%[font=宋体]乙醇[/font][font=Symbol][/font]4[font=宋体]℃冰箱过夜,醇沉多糖[/font][font=Symbol][/font]5000r/min[font=宋体]离心去上清液[/font][font=Symbol][/font][font=宋体]粗多糖[/font][font=Symbol][/font][font=宋体]脱蛋白[/font][font=Symbol][/font][font=宋体]脱色素[/font][font=Symbol][/font][font=宋体]透析[/font][font=Symbol][/font][font=宋体]浓缩[/font][font=Symbol][/font][font=宋体]冷冻干燥得水溶多糖[/font][font=Symbol][/font]DEAE-[font=宋体]纤维素色谱柱[/font][font=Symbol][/font][font=宋体]收集多糖组分[/font][font=Symbol][/font][font=宋体]测定每[/font]50s[font=宋体]洗脱液的[/font]OD[font=宋体]值[/font][font=Symbol][/font][font=宋体]绘制横坐标为管数与纵坐标为[/font]OD[font=宋体]值的洗脱曲线[/font][font=宋体]根据洗脱曲线收集多糖溶液[/font][font=Symbol][/font]4[font=宋体]℃无水乙醇醇沉过夜[/font][font=Symbol][/font][font=宋体]离心[/font][font=Symbol][/font][font=宋体]多糖沉淀[/font][font=Symbol][/font][font=宋体]常规干燥[/font][font=Symbol][/font][font=宋体]纯度鉴定[/font][font=Symbol][/font][font=宋体]纯多糖[/font]2.[font=宋体]脱蛋白[/font][font=宋体]由于粗多糖中含有一定量游离的和结合的蛋白,为保证多糖的纯度,必须将蛋白质脱去。常用的脱蛋白方法有[/font]Sevag[font=宋体]法、三氯乙酸法和蛋白酶法(如链蛋白酶、胰蛋白酶、木瓜蛋白酶)等。[/font][font=宋体]([/font]1[font=宋体])[/font]Sevag[font=宋体]法[/font][font=宋体]根据蛋白质在有机溶剂(如氯仿)中易发生变性析出的特点[/font][font=宋体],把多糖配制成[/font]5%[font=宋体]的糖液,然后按照[/font]1:3[font=宋体]的体积比加入[/font]Sevag[font=宋体]试剂(氯仿∶正丁醇[/font]=4[font=宋体]∶[/font]1[font=宋体]),混匀后剧烈振荡,静置分层后吸去水层与溶剂层交界处的变性蛋白质,重复操作多次,直到除尽蛋白质为止。[/font][font=宋体]([/font]2[font=宋体])三氯乙酸法[/font][font=宋体]将粗多糖配成[/font]5%[font=宋体]的糖液,加入[/font]30%[font=宋体]三氯乙酸溶液,使三氯乙酸终浓度为[/font]15%[font=宋体],在[/font]4℃[font=宋体]冰箱中放置过夜,离心弃去沉淀,得无蛋白的多糖溶液,再用[/font]1 mol/L NaOH[font=宋体]溶液中和至[/font]PH[font=宋体]为[/font]7[font=宋体]。[/font][font=宋体]([/font]3[font=宋体])蛋白酶法[/font][font=宋体]将粗多糖配成[/font]5%[font=宋体]的糖液,用酸碱调[/font]pH[font=宋体]至中性,加链蛋白酶[/font]0.5 L[font=宋体],放恒温箱中[/font]37℃[font=宋体]保温[/font]24 h[font=宋体]后,加热升温至[/font]80℃[font=宋体]使酶失活,离心,得无蛋白的多糖溶液。[/font]3.[font=宋体]脱色[/font][font=宋体]([/font]1[font=宋体])[/font] [font=宋体]活性炭法[/font][font=宋体]将脱蛋白后的多糖配成[/font]5%[font=宋体]的糖液,用[/font]NaOH[font=宋体]溶液调[/font]pH[font=宋体]为[/font]4.5[font=宋体],加入活性炭粉末,于[/font]80℃[font=宋体]保温[/font]2 h[font=宋体]后过滤。反复进行[/font]3[font=宋体]次,直到溶液颜色不再降低为止。[/font][font=宋体]([/font]2[font=宋体])过氧化氢法[/font][font=宋体]将脱蛋白后的多糖溶液,用浓氨水调至[/font]pH[font=宋体]为[/font]8.0[font=宋体],逐滴滴加[/font]20%H[sub]2[/sub]O[sub]2[/sub][font=宋体]至溶液为浅黄色,[/font]50℃[font=宋体]水浴,保温[/font]2 h[font=宋体]后过滤。[/font]4.[font=宋体]透析脱盐[/font][font=宋体]将脱蛋白、脱色后多糖配成[/font]5%[font=宋体]溶液,置于透析袋中,先用自来水逆向流水透析[/font]72 h[font=宋体]后,再用蒸馏水透析[/font]24 h[font=宋体],每[/font]4 h[font=宋体]换水一次。[/font]5.[font=宋体]干燥[/font][font=宋体]将脱蛋白、脱色、脱盐后的中草药多糖溶液,水浴[/font]80℃[font=宋体]浓缩至[/font]10 mL[font=宋体],加三倍体积的无水乙醇过夜,离心取沉淀,经无水乙醇、乙醚洗涤后,常规干燥得中草药去蛋白去色素的多糖。[/font]6.[font=宋体]多糖含量测定[/font][font=宋体]采用苯酚[/font]-[font=宋体]硫酸法测定样品中的多糖含量。绘制标准曲线,根据葡萄糖标准曲线和样品吸光值计算其糖含量。[/font][font=宋体]标准曲线的制作:准确称取[/font]105℃[font=宋体]干燥恒重的标准葡萄糖[/font]50 mg[font=宋体]定容于[/font]500 mL[font=宋体]容量瓶中,加水至刻度,用移液管分别吸取[/font]0[font=宋体]、[/font]0.1[font=宋体]、[/font]0.2[font=宋体]、[/font]0.3[font=宋体]、[/font]0.4[font=宋体]、[/font]0.5[font=宋体]、[/font]0.6[font=宋体]、[/font]0.7[font=宋体]、[/font]0.8[font=宋体]、[/font]0.9[font=宋体]、[/font]1.0 mL[font=宋体]转入试管中,各以蒸馏水补至[/font]1.0 mL[font=宋体],每个浓度重复四个平行。然后分别向每支试管中加入[/font]4%[font=宋体]苯酚试剂[/font]1mL[font=宋体]及浓硫酸[/font]2 mL[font=宋体],摇匀,沸水浴中煮沸[/font]10 min[font=宋体],冷却至室温,放置[/font]10 min[font=宋体]后在[/font]λ= 480 nm[font=宋体]处测定吸光值[/font]A[font=宋体]。以吸光值[/font]A[font=宋体]为纵坐标,糖含量[/font]C[font=宋体]为横坐标,得糖的标准曲线。[/font][font=宋体]样品溶液制备:精密称取水溶性粗多糖[/font]5 mg[font=宋体],先加[/font]10 mL[font=宋体]蒸馏水溶解,然后定容至[/font]50 mL[font=宋体]。[/font][font=宋体]样品含量测定:用移液管吸取样品液[/font]1.0 mL[font=宋体],加入[/font]4%[font=宋体]苯酚溶液[/font]1 mL[font=宋体]及浓硫酸[/font]2 mL[font=宋体],按上述方法进行操作,测其吸光值,以标准曲线计算样品糖含量[/font][font=宋体]按下式计算中草药多糖的质量浓度与得率。[/font] [img=,300,41]https://ng1.17img.cn/bbsfiles/images/2021/11/202111021041176368_5305_3528941_3.gif!w300x41.jpg[/img]7.[font=宋体]分级[/font][font=宋体]([/font]1[font=宋体])[/font] DEAE-[font=宋体]纤维素的预处理和装柱[/font][font=宋体]将[/font]DEAE-[font=宋体]纤维素用蒸馏水充分浸泡溶胀后,倾倒除去水分,再用[/font]0.5 M NaOH[font=宋体]溶液浸泡[/font]1 h[font=宋体],用蒸馏水反复洗涤至[/font]PH[font=宋体]等于[/font]7[font=宋体]。再用[/font]0.5 M[font=宋体]的[/font]HCl[font=宋体]溶液浸泡[/font]1 h[font=宋体],用蒸馏水洗至中性,真空清除气泡,备用。[/font][font=宋体]装柱时,先将[/font]DEAE-[font=宋体]纤维素沿着玻璃棒缓慢倒入,以防产生气泡。柱子装好后,依次用[/font]5[font=宋体]倍柱体积的蒸馏水、[/font]3[font=宋体]倍柱体积的[/font]1.0 M NaCl[font=宋体]和[/font]5[font=宋体]倍柱体积的蒸馏水进行平衡,流速设定为[/font]20 cm/ h[font=宋体]。[/font][font=宋体]([/font]2[font=宋体])水溶性粗多糖的离子交换柱层析的线性梯度洗脱[/font][font=宋体]称取[/font]50 mg[font=宋体]水溶性粗多糖,溶于[/font]5 mL[font=宋体]蒸馏水中,在已平衡好的[/font]DEAE-[font=宋体]纤维素离子交换柱([/font]1.5 × 14 cm[font=宋体],[/font]Cl[sup]-[/sup][font=宋体]型)上进行上样,先用[/font]100 mL[font=宋体]蒸馏水做流动相进行洗脱,再用[/font]0~1.0 M NaCl[font=宋体]溶液[/font]300 mL[font=宋体]进行线形梯度洗脱,流速[/font]1.0 mL/min[font=宋体],每个试管收集[/font]3 mL[font=宋体]洗脱液,苯酚—硫酸法测定洗脱液中相对的糖含量分布。[/font]8.[font=宋体]纯度鉴定[/font][font=宋体]([/font]1[font=宋体])高效液相色谱法[/font][font=宋体]采用高效液相色谱仪系统,[/font] 10A[font=宋体]检测器,[/font]CLASS-Vp[font=宋体]工作站,[/font]TSK-gel G-3000PWXL[font=宋体]不锈钢色谱柱([/font]7.8 × 300 mm)[font=宋体],柱温为[/font]40℃[font=宋体]。[/font][font=宋体]取各级多糖样品溶于[/font]0.7 M Na2SO4[font=宋体]溶液中,其多糖终浓度为[/font]2 mg/mL[font=宋体],上样[/font]20 μL[font=宋体],流动相为[/font]0.7 M Na2SO4[font=宋体]溶液,流速为[/font]0.5 mL/min[font=宋体]。[/font][font=宋体]([/font]2[font=宋体])紫外光谱分析[/font][font=宋体]将水溶性粗多糖各级分多糖样品配成[/font]0.1 mg/mL[font=宋体]的溶液,用[/font]752PC[font=宋体]型紫外可见分光光度计于[/font]200-400 nm[font=宋体]处扫描检测。[/font][font=宋体]([/font]3[font=宋体])旋光仪分析[/font][font=宋体]不同的多糖具有不同的比旋度,它们在不同浓度的乙醇中具有不同溶解度,所以,如同一多糖水溶液经不同浓度的乙醇沉淀所得的沉淀物,具有相同比旋度,则证明该多糖为均一组分。[/font][font=宋体]将评估多糖样品溶于水,成为近似半饱和溶液,置于磁力搅拌器上,在搅拌下加乙醇,使溶液中乙醇浓度达到[/font]20%[font=宋体],搅拌片刻,使沉淀完全,离心得沉淀。上清液中再继续滴加乙醇,使溶液中乙醇浓度达[/font]40%[font=宋体],所产生的沉淀再经离心。前后两次沉淀,分别干燥后在相同条件下测定其水溶液的比旋度。[/font][font=宋体]将[/font]10 mL5%[font=宋体]饱和糖液用国产[/font]WXG[font=Symbol]-[/font]6[font=宋体]型自动旋光仪,钠灯光源,以蒸馏水调零点,[/font]1dm[font=宋体]管室温下测定。[/font]
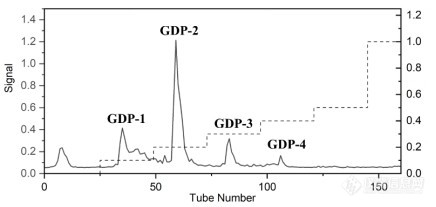
[align=center][b][font=Arial][font=宋体]天然多糖研究体积排阻色谱柱选择初探[/font][/font][/b][/align][font=Arial][font=宋体]多糖是最丰富的生物聚合物,已被发现参与许多生物过程,例如细胞间通讯、胚胎发育、细菌和[/font]/[font=宋体]或病毒感染以及体液和细胞免疫。因此,多糖与核苷酸、蛋白质、脂质一起构成了生命科学中最重要的四大生物大分子。[/font][/font][font=Arial][font=宋体]尽管多糖已在各种工业应用中使用了几十年,例如[/font] [font=宋体]药物、生物材料、食品和营养以及生物燃料,对多糖在生命科学中的重要性的日益了解和深入研究正在推动多糖用于新型(生物分子)应用的开发。不断有来自植物(膳食纤维、草药和木本植物)、藻类和地衣的生物活性多糖,以及来自动物的其他生物活性多糖(例如肝素、硫酸软骨素和透明质酸)被报道。[/font][/font][font=Arial][font=宋体]天然多糖发现的主要方法为提取纯化得到相对较纯的多糖组分,从而进行结构表征和活性评价。具体而言,通过水提醇沉得到粗多糖,再通过弱阴离子交换色谱法([/font]DEAE[font=宋体])进行纯化,得到不同盐梯度洗脱的组分。对于这些组分,需要通过体积排阻色谱法([/font][font=Arial]SEC[/font][font=宋体])分析判定,是否需要进一步纯化;得到纯品之后,需要测定多糖组分的分子量;研究多糖在体内的降解规律,也需要[/font][font=Arial]SEC[/font][font=宋体]判定多糖分子量的变化。因此,多糖研究实验室往往需要多支[/font][font=Arial]SEC[/font][font=宋体]色谱柱,以适应不同的用途。研究人员热切期望有一只首选[/font][font=Arial]SEC[/font][font=宋体]色谱柱,可以快速开展研究。本文以小秦艽多糖的研究为例,说明[/font][font=Arial]BioCore SEC-1000[/font][font=宋体]作为多糖研究的首选色谱柱,在多个用途中的效果。[/font][/font][font=Arial][font=宋体]小秦艽,学名为达乌里秦艽[/font]([/font][i][font=Arial]Gentiana dahurica Fisch[/font][/i][font=Arial])[font=宋体],为龙胆科植物龙胆属多年生草本植物,始载于《神农本草经》,列为中品,具有祛风湿、清湿热、止痹痛、退虚热的功效。小秦艽多糖经过提取和除杂后,通过中压制备[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]([/font][font=Arial]DEAE[/font][font=宋体]填料)填料纯化,得到四个组份。在后续多糖组份纯度判定和降解规律研究中,使用了[/font][font=Arial]BioCore SEC-1000[/font][font=宋体]色谱柱。[/font][/font][b][font=Arial][font=宋体]一、实验条件与方法[/font][/font][font=Arial]1.1. [font=宋体]仪器及色谱条件[/font][/font][/b][font=Arial][font=宋体]高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]:安捷伦[/font] 1260 infinity II[font=宋体],配有[/font][font=Arial]Wyatt[/font][font=宋体],[/font][font=Arial]Optilab T-REX[/font][font=宋体]示差折光检测器([/font][font=Arial]RI[/font][font=宋体])[/font][font=Arial] [/font][font=宋体]岛津[/font][font=Arial]LC-2030C[/font][font=宋体],配有岛津[/font][font=Arial]RID-20A1.2[/font][font=宋体]示差折光检测器([/font][font=Arial]RI[/font][font=宋体]);大连依利特有限公司 [/font][font=Arial]Elite 1100 [/font][font=宋体][url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],配有岛津[/font][font=Arial]RF-20AXS[/font][font=宋体]荧光检测器([/font][font=Arial]FLD[/font][font=宋体])。[/font][/font][font=Arial][font=宋体]色谱柱:[/font]T (13μm[font=宋体],[/font][font=Arial]7.8×300mm)[/font][font=宋体]或[/font][font=Arial]Biocore SEC-1000[/font][font=宋体]([/font][font=Arial]5μm, 7.8×300mm[/font][font=宋体]);[/font][/font][font=Arial][font=宋体]流动相:[/font]50 mM NH[/font][sub][font=Arial]4[/font][/sub][font=Arial]OAc (80%)+Methanol (20%)[font=宋体];[/font][/font][font=Arial][font=宋体]流速:[/font]0.5 mL/min[font=宋体];[/font][/font][font=Arial][font=宋体]柱温:[/font]25[/font][font=宋体]℃[/font][font=Arial][font=宋体];[/font][/font][font=Arial][font=宋体]进样量:[/font]20 μL[font=宋体];[/font][/font][font=Arial][font=宋体]采集时间:[/font]30 min[font=宋体]。[/font][/font][b][font=Arial]1.2 [font=宋体]样品处理[/font][/font][/b][font=Arial]1[font=宋体])秦艽多糖组份纯度分析:称取秦艽多糖[/font][font=Arial]GDP-3[/font][font=宋体](即样品[/font][font=Arial]0.3 M[/font][font=宋体])约[/font][font=Arial]5 mg[/font][font=宋体],溶于流动相,配制成[/font][font=Arial]10 mg/mL[/font][font=宋体]溶液。上样前用[/font][font=Arial]0.22 μm[/font][font=宋体]滤膜过滤。[/font][/font][font=Arial]2[font=宋体])多糖[/font][font=Arial]GDP-2[/font][font=宋体]体外稳定性的考察: [/font][font=Arial]GDP-2[/font][font=宋体]的经 [/font][font=Arial]FITC[/font][font=宋体]荧光标记(产物为[/font][font=Arial]FGDP-2[/font][font=宋体]),进行体外模拟消化。模拟胃液由胃蛋白酶[/font][font=Arial](10 g/L)[/font][font=宋体]和稀[/font][font=Arial]HCl (16.4 mL/L)[/font][font=宋体]组成,调节[/font][font=Arial]pH[/font][font=宋体]至[/font][font=Arial]1.3[/font][font=宋体]。将[/font][font=Arial]FGDP-2 (25 mg/mL)[/font][font=宋体]与各人工胃液和人工肠液[/font][font=Arial]10 mL[/font][font=宋体]混合,[/font][font=Arial]37[/font][/font][font=宋体]℃[/font][font=Arial][font=宋体]孵育。分别于[/font]0 h[font=宋体]、[/font][font=Arial]4 h[/font][font=宋体]、[/font][font=Arial]6 h[/font][font=宋体]、[/font][font=Arial]12 h[/font][font=宋体]取孵育液各[/font][font=Arial]500 μL[/font][font=宋体]作为[/font][font=Arial]HPLC-FLD[/font][font=宋体]测定样品。用[/font][font=Arial]0.2 M NaOH[/font][font=宋体]和[/font][font=Arial]TCA (20 % , w/w)[/font][font=宋体]终止反应。[/font][/font][b][font=Arial][font=宋体]二、结果与讨论[/font][/font][/b][font=Arial][font=宋体]多糖纯度及分子量的测定是多糖结构解析及构效关系研究的关键步骤。小秦艽多糖的纯化色谱洗脱曲线如图[/font]1[font=宋体]所示,一共得到四个组份。[/font][/font][align=center][img=,348,]https://ng1.17img.cn/bbsfiles/images/2023/02/202302081451227890_6619_3237657_3.jpg!w436x207.jpg[/img][/align][align=center][font=Arial][font=宋体]图[/font]1 [font=宋体]小秦艽多糖的[/font][font=Arial]DEAE[/font][font=宋体]纯化色谱图(硫酸苯酚法重构)[/font][/font][/align][font=Arial][font=宋体]组份经透析、冻干后,首先需要确定它的纯度。如果组份不纯,则后续的核磁分析将是徒劳无功的。我们实验室前期配备了三支不同孔径的[/font]SEC[font=宋体]色谱柱,经分析组份[/font][font=Arial]1[/font][font=宋体]和[/font][font=Arial]2[/font][font=宋体]较纯,而[/font][font=Arial]3[/font][font=宋体]和[/font][font=Arial]4[/font][font=宋体]不纯。如果不纯,则需要通过凝剂渗透色谱([/font][font=Arial]GPC[/font][font=宋体])进一步纯化。[/font][font=Arial]SEC[/font][font=宋体]分析可以确定组份的数目,以在[/font][font=Arial]GPC[/font][font=宋体]纯化时收集馏分。[/font][/font][font=Arial][font=宋体]在用实验室已有[/font]SEC[font=宋体]色谱柱分析时(图[/font][font=Arial]2[/font][font=宋体]上),色谱峰数目不明确。该色谱柱孔径为[/font][font=Arial]450?[/font][font=宋体],粒径为[/font][font=Arial]13 μm[/font][font=宋体],分离度不理想,可能分子量超出其适用范围。于是改用孔径更大且粒径为[/font][font=Arial]5 μm[/font][font=宋体]的[/font][font=Arial]Biocore SEC-1000[/font][font=宋体]柱进行试验(图[/font][font=Arial]2[/font][font=宋体]下),结果表明分离效果好,明显识别为三个色谱峰。为该组份的后续纯化以及多糖稳定性的考察提供基础。[/font][/font][align=center][img=,264,]https://ng1.17img.cn/bbsfiles/images/2023/02/202302081451353696_6101_3237657_3.jpg!w331x346.jpg[/img][/align][align=center][font=Arial][font=宋体]图[/font]2 [font=宋体]秦艽多糖分子量的分析色谱图[/font][/font][/align][align=center][font=Arial][font=宋体](上图,实验室已有色谱柱[/font]T[font=宋体];下图,纳谱[/font][font=Arial]Biocore SEC-1000[/font][font=宋体])[/font][/font][/align][font=Arial][font=宋体]此外,该色谱柱,分离中等分子量多糖标准品([/font]36 kDa ~ 131 kDa[font=宋体])时,也呈现了较好的线性关系。因此,我们认为该色谱柱具有分子量适用范围宽的优点,可以作为多糖分析中的通用[/font][font=Arial]SEC[/font][font=宋体]色谱柱。[/font][/font][font=Arial][font=宋体]为了确定秦艽多糖的体外消化模式,选取了经测量相对较纯的秦艽多糖[/font]GDP-2[font=宋体]组分进行荧光标记,建立了[/font][font=Arial]HPLC - FLD[/font][font=宋体]分析方法,进行体外稳定性考察。通过上述实验对[/font][font=Arial]SEC[/font][font=宋体]柱的比较分析,选取了分离效果较好的[/font][font=Arial]Biocore SEC-1000[/font][font=宋体]进行该实验。[/font][font=Arial]FGDP-2[/font][font=宋体]在模拟胃液中的分子量随时间变化色谱图如图[/font][font=Arial]3[/font][font=宋体]所示,发现色谱峰的相对峰高向小分子偏移,说明[/font][font=Arial]FGDP-2[/font][font=宋体]在消化[/font][font=Arial]4 h[/font][font=宋体]后发生部分降解。可能是由于多糖对酶和强酸敏感,导致多糖的糖苷键断裂。然而,观察到整个消化系统中没有单糖出现,胃和肠道不会造成多糖结构的过度损失。[/font][/font][align=center][img=,288,]https://ng1.17img.cn/bbsfiles/images/2023/02/202302081451479308_4610_3237657_3.jpg!w361x212.jpg[/img][/align][align=center][font=Arial][font=宋体]图[/font]3[font=宋体]模拟胃液消化过程中[/font][font=Arial]FGDP-2[/font][font=宋体]的[/font][font=Arial]HPLC - FLD[/font][font=宋体]表征[/font][/font][/align][font=Arial][font=宋体]三、小结[/font][/font][font=Arial][font=宋体]多糖研究人员往往面对不同来源的多糖分子,分子量差异较大,从几万到上百万的多糖分子量。在多糖的研究过程中,需要一支通用的[/font]SEC[font=宋体]色谱柱,进行快速分子量判断,以确定后续研究操作。[/font][font=Arial]BioCore SEC-1000[/font][font=宋体]在我们对小秦艽多糖的分子量纯度分析、分子量测定和体外消化稳定性等研究中,均获得了良好的效果。我们认为,[/font][font=Arial]BioCore SEC-1000[/font][font=宋体]可以作为多糖研究实验室的首选[/font][font=Arial]SEC[/font][font=宋体]色谱柱。[/font][/font][font=Arial] [/font][font=Arial][font=等线]公司:[/font][/font][font=等线][font=等线]苏州大学药学院[/font][/font][font=等线][font=等线]色谱柱信息:[/font][/font][font=Arial]BioCore SEC-1000 5μm, 7.8×300mm[/font][font=宋体] [/font]
分离纯化谷物β-葡聚糖一般用什么阴离子交换色谱柱和凝胶过滤色谱柱哇
请教如何出去多糖中的单糖和双糖
[color=#333333]该文建立了大孔树脂-高速逆流色谱分离中药材地黄中有效成分毛蕊花糖苷的方法。考察了4种大孔树脂对地黄粗提物中毛蕊花糖苷的静态吸附与解吸情况,其中D101大孔树脂对目标成分的吸附率与解吸率最理想,实验结果表明体积分数为10%的乙醇洗脱得到的毛蕊花糖苷含量最高,目标成分含量从4.9%提高到32.6%。最后,部分纯化的样品(165 mg)采用高速逆流色谱进一步纯化,两相溶剂系统由乙酸乙酯-正丁醇-水(1∶4∶5,v/v/v)组成,分离得到45 mg纯度为96%的毛蕊花糖苷。 [/color]
最近在做生姜糖蛋白的提取纯化,看了很多文献,都要用到DEAE-52纤维素纯化,想问一下大家收集管数应该按照什么计算,是按照柱体积吗,多少个柱体积为一管呢?目前洗脱条件如下:上样浓度50mg/ml,上样量1ml,柱子规格16mm×20cm,洗脱液0.2mol/L Tris缓冲液(含0.1mol/LNaCl),pH7.8,每10ml一管进行收集,共收集50管,在1-5管处有蛋白和多糖共同的洗脱峰。
想请教一下大家,像deae纤维素柱和葡聚糖凝胶柱纯化多糖,能洗脱下来的组分,一般几个柱体积可以洗脱得差不多?
我现在正在做植物中多糖的提取、分离和纯化,在测定多糖分子量时,若没有除蛋白会不会影响结果?还有多糖要多纯才能进行测定分子量呢?谢谢
[em0809] 大家好,我提取的多糖想上液相前纯化一下,买的酸性氧化铝针筒式预柱,但是不知道活化,洗涤,洗脱液都用什么,那些原理我都看了,只是不知道具体到操作步骤怎么弄, 哪位帮帮忙啊,谢谢[em0808]
[color=#444444]我用我们实验室的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]来测多糖结构,但是我们的柱子是测醇类物质的,不知道可不可以直接用,若是可以的话我应该做哪些前期处理呢?我想是不是可以把糖分解成醇类物质后再走柱子呢?还请各位高手指点一二,我想测多糖结构应该都做哪些呢?谢谢大家的慷慨帮助[/color]
能否请教下用液相色谱来检测多糖及多糖的氧化和还原态的方法?另外我对液相色谱仪的使用还不是特别了解,希望能知道更多有关仪器使用的方法和其需要的注意事项。谢谢!
多糖类手性固定相在色谱中的应用郑 芸 方积年(中国科学院上海生命科学院上海药物研究所,上海201203)摘要 手性色谱技术是最重要的手性分离方法之一,它不仅可以快速地分析对映体纯度,也可以用于大量制备光学异构体。设计和发展高效的固定相是手性色谱技术的核心。在诸多的手性固定相中,多糖类手性固定相因品种繁多、耐用而被广泛应用。本文综述了多糖类手性固定相在高效液相色谱、模拟移动床色谱、超临界流体色谱及膜分离中的应用。共引用文献52篇。关键词 多糖,手性固定相,色谱,评述1 引 言 近20年来,用色谱方法分离手性化合物取得了显著进展,已广泛应用于许多领域,如药物化学、不对称合成和生物分析等,不仅可以测定光学纯度,也可用于大量制备光学异构体。 手性色谱技术的核心是设计和制备适用范围广的手性固定相(chiral stationary phase,CSP)。至今已制备出大量用于色谱的CSP,其中120多种已商品化。CSP可分为两大类:一类是由小分子固定在硅胶载体上构成(刷型或Pirkle型),另一类是用光学聚合物固定在载体上制成,多孔胶状的聚合物也可直接用作CSP。其中Okamoto等发展的多糖类固定相是非常有用的分离工具,它们种类繁多、耐用而且负荷量大。其它广泛使用的手性固定相有衍生化的酒石酸CSP(Kromasil—TBB) ]、a1一酸性糖蛋白、Pirkle固定相、环糊精、聚丙烯酰胺和大环抗生素,如万古霉素、teicoplanin和瑞斯托菌素以及最新的用分子印记技术及仿生传感技术发展的CSP 。 多糖,如纤维素和淀粉是自然界大量存在的有光学活性的生物聚合物。它们具有良好的精细结构,能拆分异构体,包括氨基酸衍生物和联苯衍生物的阻转异构体,但它们的手性识别能力不强,适用面也很窄,只能用于毛细管电泳(CE)分析中。半合成的经过改性的多糖适用范围则大大扩展,可用于LC、CE、SFC、TLC、膜分离及萃取中,既可用于分析也可用于制备。经研究发现,多糖类衍生物的手性识别能力与单糖残基的性质、连接位置和连接形式有关。2 高效液相色谱(HPLC) 多糖类手性固定相在HPLC中的应用相当广泛,常见的商品化多糖类手性固定相及应用实例可参考相关文献。纤维素类多糖为刚性的线形结构,而淀粉类多糖具有螺旋形结构。据报道有84%的小分子外消旋化合物可用Chiralcel OJ、Chiralcel OD、Chiralpak AD、Chiralpak AS分离 。用HPLC分析对映体时,除了常用的UV或示差折光指数检测器,还可使用专门检测手性物质的旋光检测器和圆二色散检测器。这也是HPLC比[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和NMR光谱等其它分析方法的优越之处。 为提高手性分离效果以利于检测,还可以对样品进行适当的衍生化。Fukushima 用荧光试剂DBD—PZ([4.[(N,N—dimethylamino)一sulfony1]-7一piperazino-2,1,3-benzoxadiazole])和DBD—COHz(4[[(N—hydrazinoformy1)methy1]一N—methy1]amino-7一[N,N一(dimethylamino)sulfony1]-2,1,3-benzoxadiazole)对(RS)-2一芳基丙酸类化合物进行了衍生化,并发现衍生物洗脱顺序发生改变。3 动态高效液相色谱(DHPLC) 新发展的手性DHPLC方法 可用于研究高温时立体化学稳定的手性化合物,它可得到一系列受温度控制的平顶或峰形曲线,从而可以考察对映体互变的动态过程、动力学数据及对映体互变的能垒。一般在CSP上用色谱方法分离外消旋混合物,最多可以得到收率50% 的两种纯的光学异构体。而在DHPLC中,利用CSP来达到对映体互变平衡,从而使分离和平衡合二为一,理论上可以从外消旋混合物中以100%收率得到一种纯的光学异构体。它的基本原理是外消旋混合物立体化学稳定在较低温度时对映体互变过程被抑制,而较高温度发生对映体互变。实验中让外消旋混合物先通过一个低温CSP柱子,将先洗脱出来的组分(A)继续通人第二个高温CSP柱子,收集后洗脱组分(B)。(A)进入第二个柱子后停留足够长时间达到对映体互变平衡,再继续洗脱,得到(A)和(B)。如果进行多次循环平衡、过柱,则可得到纯的对映体(B)。如Lorenz等用DHPLC分离一螺环化合物,该化合物可通过螺环处的C—O 键开环和闭环进行对映体互变。将它依次通过0℃和40℃ 的两根Chiralcel OD柱,平衡2h,即可得到32% ee(enantiomerie excess,ee)的(+)一对映体。4 模拟移动床色谱(SMB) 至今批次处理色谱在应用中仍占主导地位,但大规模制备需要大量CSP。CSP价格昂贵,而且产品的浓度低,洗脱液消耗量大,难以回收。SMB可以节省90% 的流动相并得到更高的产率。在批次处理色谱中被分离组分在流动相的驱动力下移动,固定相只有一小部分起作用。在移动床色谱中,不仅流动相发生移动,固定相也要向相反方向移动,易洗脱的化合物(萃余液)随流动相移动,难洗脱的化合物(萃取液)随固定相移动。整个固定相的分离能力被持续利用,明显地提高了系统产率。但就技术而言很难移动固定相,因此采用模拟方式,SMB的环状柱子实际上是用许多小柱依次连接而成,有规律地改变进样口和出样口,可以达到和固定相移动相同的效果。SMB技术起于20世纪60年代UOP(Universal Oil Products,Des Plaines,IL,USA)从C8 中分离对二甲苯,后来该技术被广泛用于制药工业,以获得光学纯药物。其中应用于SMB的CSP约有70%是多糖类CSP。如Nagamatsu等用SMB方法替代以前的非对映体结晶的方法,用稍做改性的Chiralcel OF(cellulose 4-chlorophenyl carbamate)分离了一种制药工业的中间体喹啉甲瓦龙酸酯。Francotte等的研究还发现,SMB对于难溶的化合物,如formoterol尤为有用。而且可调节不同参数如进样率和萃取率来达到最佳纯度和产率。
采用水提醇法提取天麻粗多糖,用Sevag法除去蛋白质,用CTAB法纯化,得到纯化后的天麻多糖。水解得到的单糖,分别有5个样品。
凝胶过滤色谱摘要:本文主要讲解了凝胶过滤色谱法(分子筛)在蛋白纯化实验中的应用,包括纯化原理、实验方案设计、技术操作以及相关案例介绍和问题分析。基本原理凝胶过滤色谱蛋白纯化法,又称为空间排阻色谱,分子筛等。其原理是应用蛋白质分子量或分子形状的差异来分离。当样品从色谱柱的顶端向下运动时,大的蛋白质分子不能进入凝胶颗粒从而被迅速洗脱;而较小的蛋白质分子能够进入凝胶颗粒中,且进入凝胶的蛋白在凝胶中保留时间也不同,分子量越大,流出时间就越早,最终分离分子大小不同的蛋白质。http://www.detaibio.com/assets/image/topics/gel-filtration-chromatography-theory.jpg通常,多数凝胶基质是化学交联的聚合物分子制备的,交联程度决定凝胶颗粒的孔径。常用的色谱基质有:葡聚糖凝胶(Sephadex)、琼脂糖凝胶(Sepharose)、聚丙烯酰氨凝胶(Bio-Gel P)等。高度交联的基质可用来分离蛋白质和其他分子量更小的分子,或是除去低分子量缓冲液成分和盐,而较大孔径的凝胶可用于蛋白质分子之间的分离。选用合适孔径的凝胶很大程度取决于目标蛋白的分子量和杂蛋白的分子量。实验方案设计凝胶介质的选择凝胶介质的选择主要是根据待分离的蛋白和杂蛋白的分子量选择具有相应分离范围的凝胶,同时还需要考虑到分辨率和稳定性的因素。比如,如果是要将目的蛋白和小分子物质分开,可以根据他们分配系数的差异,选用Sephadex G-25和 G-50;对于小肽和低分子量物质的脱盐,则可以选用Sephadex G-10、G-15以及Bio-Gel P-2或P-4;如果是分子量相近的蛋白质,一般选用排阻限度略大于样品中最高分子量物质的凝胶。具体凝胶过滤色谱介质应用如下:常用凝胶过滤色谱介质的分离范围凝胶介质蛋白质的分离范围/103凝胶介质蛋白质的分离范围/103Sephadex G25Sephadex G50Sephadex G100Sephadex G200Sepharose 6BSepharose 4B1~51.5~304~1505~60010~400060~20000Sepharose 2BBio-Gel P-4Bio-Gel P-10Bio-Gel P-60Bio-Gel P-150Bio-Gel P-30070~400000.5~45~1730~7050~150100~400凝胶介质的预处理凝胶在使用前应用水充分溶胀(胶:水=1:10),自然溶胀的耗时较长,可采用加热的方法使溶胀加速,即在沸水浴中将凝胶升温至沸,1~2h即可达到溶胀。在烧杯中将干燥凝胶加水或缓冲液,搅拌,静置,倾去上层混悬液,除去上清液中的凝胶碎块,重复数次,直到上清澄清为止。色谱柱的选择色谱柱的体积和高径比与色谱分离效果密切相关,凝胶柱床的体积、柱长和柱的直径以及柱比的选择必须根据样品的数量,性质和分离目的进行确定。组别分离时,大多采用2~30cm长的色谱柱,柱床体积为样品溶液体积的5倍以上,柱比一般在5~10之间;而分级分离一般需要100cm左右的色谱柱,并要求柱床体积大于样品体积25倍以上,柱比在20~100之间。凝胶柱的填装凝胶色谱柱与其它色谱方法不同,溶质分子与固定相之间没有力的作用,样品组分的分离完全依赖于他们各自的流速差异。装住时关住柱子下口,在柱内加入约1/3柱床体积的水或缓冲液,然后沿着柱子一侧将缓冲液中的凝胶搅拌均匀,缓慢并连续的一次性注入柱内。待凝胶沉积约5厘米左右时,打开柱子下口,控制流速在1ml/min。样品的处理与上样根据样品的类型和纯化分析,需要选择合适的缓冲液,为了达到良好的分析效果,上样量必须保持在较小的体积,一般为柱床体积的1%~5%,蛋白质样品上样前应进行浓缩,使样品浓度不大于4%(样品浓度与分配系数无关),但需要注意的是,较大分子量的物质,溶液粘度会随浓度增加而增大,使分子运动受限,影响流速。上样前,样品要经滤膜过滤或离心,除去可能堵塞色谱柱的杂质。洗脱与收集凝胶过滤色谱的缓冲液用单一缓冲液或含盐缓冲液作为洗脱液即可,主要考虑俩个方面的原因:蛋白的溶解性和稳定性。所用的缓冲液要保证蛋白质样品在其中不会变性或沉淀,PH应选在样品较稳定、溶解性良好的范围之内,同时缓冲液中要含有一定的盐(NaCL),对蛋白质起稳定和保护作用。洗脱过程中始终保持一定的操作压,流速不可过高,保持在0.5~3.0mL/min即可。案列介绍AKTA凝胶过滤色谱分离蛋白质材料色谱介质:Sephacryl S-200,蛋白质分离范围(5~250)×103 色谱柱:XK16/60预装柱色谱设备:AKTA Explorer混合样品:含单克隆抗体,分子量180000;牛血清白蛋白(68000),溶菌酶(14000)NaOH 0.5 mol/LNaCl 200 mmol/LPB 20 mmol/LPH7.0 缓冲液方法凝胶除菌处理超纯水冲洗柱子后,用0.5mol/L NaOH正向冲洗柱,流速3mL/min,冲洗3柱体积平衡NaOH处理完毕后,用超纯水冲洗2柱体积,接着用含200mmol/L NaCl和20mmol/L PB的7.0PH缓冲液冲洗5~10倍柱体积上样平衡完毕后,选择样品泵进行上样,上样流速3mL/min,上样体积为1mL洗脱上样结束后,用平衡缓冲液进行洗脱清洗与保存纯化结束后,用0.5mol/L NaOH反向冲洗2柱体积,冲洗时间30~60min,冲洗结束后,用超纯水正向冲洗5柱体积,再用20%乙醇冲洗3柱体积,然后拆下柱子,俩端封死,低温保存。问题分析和解决方案色谱分离前如何净化样品在色谱分离前,对样品进行离心和过滤,离心能除去大部分块状物,如果离心后样品仍不清澈,可用滤膜过滤。由醋酸纤维薄膜或PVDF材料制成的滤膜能够非特异性的结合少量蛋白。溶液交换不彻底严格控制上样体积,上样体积不超过柱体积30%。若样品溶液体积较多,可以分多次上样,注意每次上样时间间隔,可根据电导色谱峰确定下一次上样时间。分辨率不高1)提高装柱质量,使色谱柱装填匀实; 2)提高柱床高度; 3)控制上样体积,最大上样体积不超过柱床体积5%; 4)控制样品黏度与洗脱溶液黏度保持一致; 5)根据样品特点选择合适的洗脱溶液,调节洗脱溶液的离子强度或亲水性; 6)选择合适的凝胶柱(如何选择请参照上文)色谱峰对称性差1)提高装柱质量,装柱匀实——若柱装的太松,容易引起拖尾,装的太紧,会引起前沿;2)柱较脏,再生色谱柱肩峰出现的原因及解决方法1)柱床松动,重新装柱或反向冲洗柱2)柱筛板堵塞,超声清洗筛板3)柱干裂,重新装柱
如题,我想用液相测提取的多糖里的单糖,我看文献上都是提取纯化后,用1-苯基-3-甲基-5-吡唑啉酮(PMP)进行衍生化后再测定,我看别人用的柱子有XDB-C18,ODS-VP色谱柱,Wondasil C18,检测器都在用紫外或者二极管阵列检测器。现在我们有一个ODS-3(C18)的柱子,不知道能不能测衍生化后的糖呢?对柱子这块我不是很懂,烦请有经验的前辈交流下,不胜感激。祝大家新年快乐~
请问有没有做过利用Thermo 的SEC 色谱柱测多糖分子量或者分离多糖的?可行么?有什么注意的地方?
[font=宋体]蛋白质是包括人类在内的各种生物有机体的重要组成成分,是生命的物质基础之一。生物体的生长、发育、遗传和繁殖等一切生命活动都离不开蛋白质。[/font][font=宋体] [/font][font=宋体]随着分子生物学、结构生物学、基因组学等研究的不断深入,人们意识到仅仅依靠基因组的序列分析来试图阐明生命活动的现象和本质是远远不够的。只有从蛋白质组学的角度对所有蛋白质的总和进行研究,才能更科学地掌握生命现象和活动规律,更完善地揭示生命的本质。[/font][font=宋体] [/font][font=宋体]由此许多学者将生命科学领域的研究焦点从基因转向蛋白质,使蛋白质成为揭示生命活动现象和分子生物学机理的重要研究对象。研究蛋白质首要的步骤是将目的蛋白从复杂的大分子混合物中分离纯化出来,得到高纯度具有生物学活性的目的物。因此,高效的纯化技术和手段是蛋白质研究的重要基础和关键之一。[/font][font=宋体] [/font][font=宋体] [/font][b][font=宋体][font=宋体]蛋白纯化的目的[/font] [/font][/b][font=宋体][font=宋体]蛋白纯化的目的是将目标蛋白质从细胞裂解液的全部组分中分离出来,同时仍保留蛋白的生物学活性及化学完整性。蛋白质的分离和提纯工作是一项艰巨而繁重的任务,需根据蛋白的特性选择合适的纯化方法来提高获得的蛋白制品的纯度。[/font] [/font][font=宋体] [/font][b][font=宋体][font=宋体]蛋白纯化的原理[/font] [/font][/b][font=宋体][font=宋体]不同蛋白质的氨基酸序列及空间结构不同,导致其在物理、化学、生物学等性质上存在差异,利用待分离蛋白质与其它蛋白质性质上的差异,即可以设计出一套合理的蛋白纯化方案。蛋白的纯化大致分为粗分离阶段和精细纯化阶段两个阶段。粗分离阶段主要将目的蛋白和其他细胞成分如[/font] [font=Calibri]RNA[/font][font=宋体]、[/font][font=Calibri]DNA [/font][font=宋体]等分开,常用的方法为硫酸铵沉淀法。精细纯化阶段的目的是把目的蛋白与其他大小及理化性质接近的蛋白区分开来,[/font][/font][b][font=宋体][font=宋体]常用的方法有:凝胶过滤层析、离子交换层析、疏水层析、亲和层析等。[/font] [/font][/b][font=宋体] [/font][b][font=宋体]①[/font][font=宋体]凝胶过滤层析[/font][/b][font=宋体]凝胶过滤层析(又叫做分子筛)是根据样品的分子大小对样品进行分离的一种简单温和的层析技术。凝胶过滤层析也称分子筛层析、排阻层析,是利用具有网状结构的凝胶的分子筛作用,根据被分离物质的分子大小不同来进行分离。不同于离子交换层析和亲和层析,凝胶过滤的层析样品不与层析柱料结合,因此,缓冲液成分不直接影响分辨率。[/font][font=宋体] [/font][font=宋体] [/font][font=宋体][font=宋体]原理:层析柱中的填料是球状颗粒的惰性的多孔网状结构的柱料,多是交联的聚糖[/font][font=Calibri]([/font][font=宋体]如葡聚糖或琼脂糖[/font][font=Calibri])[/font][font=宋体]类物质。在加入样品之后,样品中的小分子物质能进入球状填料内部,在柱子中停留时间较长;而大分子物质不能进入球状填料内部,停留时间较短。所以当样品经过凝胶过滤层析柱分离后,样品中的不同分子大小的物质就可以被分离开了。[/font][/font][font=宋体] [/font][font=宋体]特点:[/font][font=宋体] [/font][font=宋体]根据分子大小和形状进行分离[/font][font=宋体] [/font][font=宋体]是一种非吸附的分离方式[/font][font=宋体] [/font][font=宋体]缓冲液成分不直接影响分辨率,只需要一种缓冲液[/font][font=宋体] [/font][font=宋体]操作便捷[/font][font=宋体] [/font][font=宋体] [/font][b][font=宋体]②[/font][font=宋体]离子交换层析[/font][/b][font=宋体]离子交换层析是目前蛋白质分离纯化中应用最广泛的方法之一。[/font][font=宋体] [/font][font=宋体]原理:不同蛋白等电点差异,分子大小差异,在同一个流动相中电荷密度分布不同,电荷量不等,与具有相反电荷的离子交换介质结合强度不同,在流动相洗脱时保留时间不同,从而得以分离。[/font][font=宋体] [/font][font=宋体] [/font][font=宋体]特点:[/font][font=宋体] [/font][font=宋体]根据分子大小和等电点差异进行分离[/font][font=宋体] [/font][font=宋体]灵敏度高,重复性,选择性好,分析速度快[/font][font=宋体] [/font][font=宋体] [/font][font=宋体] [/font][b][font=宋体]③[/font][font=宋体]疏水层析[/font][/b][font=宋体]原理:疏水层析是依据蛋白质疏水性差异分离的。即根据蛋白质和疏水介质表面的疏水基团的可逆相互作用进行分离。蛋白的疏水性在高离子强度下被增强,因此在高离子强度环境中结合,通常采用降低离子强度的方式进行洗脱。独特的吸附分离模式使得疏水层析成为硫酸铵盐析后或离子交换高盐洗脱后理想的纯化方式。[/font][font=宋体] [/font][font=宋体]特点:[/font][font=宋体] [/font][font=宋体]采用了盐的水溶液作为流动相,色谱条件温和,生物大分子的活性回收率很高。[/font][font=宋体] [/font][font=宋体][font=宋体]蛋白质在[/font][font=Calibri]HIC[/font][font=宋体]操作过程中是高盐上样,低盐洗脱(高盐浓度的样品不必作处理就可直接上样)。[/font][/font][font=宋体] [/font][font=宋体]在一次色谱中可同时实现出去盐酸胍、蛋白质复性和分离三个目的。[/font][font=宋体] [/font][font=宋体][font=宋体]温度升高,蛋白质天然折叠伸展,暴露出更多内部疏水集团,使蛋白质的[/font][font=Calibri]HIC[/font][font=宋体]保留发生变化。[/font][/font][font=宋体] [/font][font=宋体]色谱填料稳定性好,盐水体系作流动相无环境污染。[/font][font=宋体] [/font][b][font=宋体]④[/font][font=宋体]亲和层析[/font][/b][font=宋体][font=宋体]原理:[url=https://cn.sinobiological.com/resource/protein-review/protein-purification-by-ac][b]亲和层析[/b][/url]是应用生物高分子与配基可逆结合的原理,将配基通过共价键牢固结合于载体上而制得的层析系统。这种可逆结合的作用主要是靠生物高分子对它的配基的空间结构的识别。常用的生物亲和关系有酶[/font][font=Calibri]-[/font][font=宋体]底物、底物类似物、抑制剂、激活剂、辅因子,抗体[/font][font=Calibri]-[/font][font=宋体]抗原,激素[/font][font=Calibri]-[/font][font=宋体]受体蛋白、载体蛋白,外源凝集素[/font][font=Calibri]-[/font][font=宋体]多糖、糖蛋白、细胞表面受体,核酸[/font][font=Calibri]-[/font][font=宋体]互补核苷酸序列、组蛋白、核酸结合蛋白等,具有高效、简单、快速的优点,是当前最为理想的分离纯化蛋白的方法。[/font][/font][font=宋体] [/font][font=宋体][font=宋体]更多详情可以参看蛋白纯化技术[/font][font=Calibri]/[/font][font=宋体]方法:[/font][font=Calibri]https://cn.sinobiological.com/resource/protein-review/protein-purification-techniques[/font][/font][font=Calibri] [/font]
多糖的分析是一个大问题啊!和大家讨论一下吧,经综合各种文献我认为多糖结构分析内容:要搞清1. 多糖的单糖组成(种类、比例)2. 每个单糖残基的D-、L-构型,吡喃环式或呋喃环式3. 羟基被取代情况(糖苷键的位置)4. 糖苷键及构型(α、β异头异构体)5. 重复单元方法:1、单糖组成:(对照品:葡萄糖、岩藻糖、半乳糖、甘露糖、木糖、阿拉伯糖、鼠李糖)a:水解: 纸层析薄层层析气相色谱(糖氰乙酸酯衍生物、糖醇乙酸酯)液相色谱(ZORBAX-NH2、HRC-NH2、RID)首选气相,灵敏度高,液相RSD、ELSD灵敏度低b:TFA酸解:气相色谱(乙酰化物)c:甲醇解:气相色谱(三甲基硅醚)2:高碘酸钠氧化和Smith降解a:每摩尔己糖基的高碘酸消耗量、甲酸释放量。(目的:判断可氧化糖基与不可氧化糖基之比例)b:Smith降解完全水解,气相分析,如有葡萄糖(表示有1-3键糖基)、甘油(有1-6或1-2糖基)、甲酸(有1-6糖基)Smith降解部分水解,说明主干糖苷键类型。3:甲基化分析(Hakrmor法)-支链分布多糖—甲基化—水解—还原得甲基化单糖醇—乙酰化得糖醇衍生物—GC-MS检测。 对照品 2,3,4,6-四甲基葡萄糖 糖苷键类型 1—2,4,6- 三甲基葡萄糖 1—32,3,4-三甲基葡萄糖 1—62,4-二甲基葡萄糖 1—3,6 4:IR图谱解析a:吡喃环式或呋喃环式α、β异头异构体5:1HNMR及13CNMR解析(构型)6:纯度检查:a: 紫外吸收光谱(280、260)b:电泳(琼脂糖电泳、聚丙烯酰胺凝胶、醋酸纤维素薄膜)c:薄层色谱(多糖不水解)7:X射线衍射,立体构型。好多啊!想和大家讨论讨论多糖的HPLC分析,我们试验室用的液相是C18柱,紫外检测器,做多糖含量及纯度检测,这样的装备够不够用呀?是不是做前必须衍生化或有其它方法,如用示差折射仪作检测器,是不是不需衍生化?多糖的HPLC分析,用得较多用HPGPC测分子量及分子量分布。一般纯多糖紫外吸收较弱,多用RID或ELSD。至于含量测定多用硫酸蒽酮比色或苯酚硫酸法。http://img.dxycdn.com/images_new/smiles/smile_angry.gif
想从动物心脏中纯化一种蛋白,用没有做过亲和色谱提取的?谢谢
高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析过程中可以纯化溶剂么?还是这是两个过程,我听师姐说就是分析过程中就会有纯化过程,有点懵
有谁知道反相色谱在合成肽分析、纯化中是如何应用的?谢谢
纯化气对[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的影响大不
请教高手,我用苯酚硫酸法测多糖为什么出现紫色,使用分子筛纯化后的。谢谢!
高手请指教!色谱柱被多糖堵了,水反冲柱不起作用,用乙腈冲压力又升高了,咋办呀?
请问有用过美国高麦氩色谱(GM100)的吗?其中有一个载气(氩气)的纯化器。有换过的吗?原装的太贵了将近3万,请问是否有可替代的厂家或者设备?Parameter is not valid.
维权声明:本文为环烯醚萜原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。老生常谈——开放ODS柱色谱在分离纯化中的应用什么是ODS? ODS是英文octadecyl silane的缩写,意思是十八烷基硅烷,是以硅胶为基质键合的C18填料,属于反相色谱。ODS柱色谱,简单地说,就是指用ODS装成的色谱柱啦。“开放”一词,是指未对色谱柱施加任何压力,让其在重力作用下洗脱;相对于开放ODS柱色谱,有中低压柱色谱,高低柱色谱等等。 我们平时所说的反相HPLC,一定程度上来说,其实也可以说是ODS柱色谱,只不过它的分离效果更好,还配备了紫外或者示差等高级地检测手段,其实质还是属于反相柱色谱。适用范围有哪些? 首先需要说明:ODS在分离纯化的过程中,起着极为巨大的作用!! 就化合物极性而言,ODS适合分离极性中等偏大的化合物类型。对于极性较小的化合物,应该考虑用硅胶、凝胶等手段进行分离纯化,绝对不要尝试ODS,唯一的后果只能是:样品全部死吸附,根本不可能洗脱!! 某些行业,前处理一般是萃取,比如环己烷/石油醚、乙酸乙酯/氯仿、正丁醇、水。水层的、正丁醇层的样品,一般都可以用ODS进行分离纯化;乙酸乙酯层的样品,极性偏大的部分可以考虑用ODS;至于环己烷层的样品,千万不要使用ODS,那将是:上样量多少,死吸附就有多少!! 就化合物分类而言,ODS对于黄酮苷类、环烯醚萜苷类、糖苷类等成分都能有一定的分离效果,也有很多成功的分离纯化实例。但是,对于苷元类化合物,则需要慎重考虑,其实还是极性大小的问题:苷元类化合物,一般极性较小。如何装柱、上样、洗脱? 装柱:新买来的ODS,用甲醇浸泡过夜后即可装柱。具体的装柱方法,跟硅胶的装柱子法是一样的,记得用甲醇装柱就行。装完以后,置换成起始流动相系统,即可投入使用了。 上样:分为两种,一种是湿法上样,另一种是拌样。湿法上样,不必多说,就是将样品溶解至一定体积(强烈建议用起始流动相溶解,但体积不可过大,太大相当于原点变大,分离度变差),然后上样;至于拌样,很多的观点表示ODS不应该拌样,我在这里指出,依我个人经验来看,拌样是可以的,尤其对于一些溶解性较差的样品,拌样后分离的效果,比湿法上样好很多,大家可以尝试。(所谓拌样,就是指将你的样品溶解,然后从柱子里面掏出小部分ODS,然后进行拌匀,注意不要过载) 洗脱过程:如果你有ODS薄层板,你可以先点板看看样品大概的极性大小。如果你没有ODS板,你拿到的是“盲样”,一般可以采取常规的梯度洗脱,如水——30%甲醇/水——50%甲醇/水——70%甲醇/水——100%甲醇,然后依据各流分样品量的大小进一步进行细分;经过常规的梯度洗脱以后,假设,你的样品集中在50%的甲醇/水部分,进行细分的时候,你就可以选择40%甲醇/水——45%甲醇/水——50%甲醇/水——100%甲醇这样的梯度。 流动相的选择:其实就是反相流动相,无非就是甲醇/水,乙腈/水,流动相中也可以加入一些其他物质。我曾经很多次加入乙酸,用来改善拖尾的现象(点ODS薄层板分析过)。至于缓冲盐,我没有试过,具体情况不太了解。洗脱样品的如何处理? 洗脱下来的样品,可用硅胶薄层板进行点板分析,然后将相同流分进行合并,也可以采用HPLC检测合并。 我认为,对于一些量小的流分,相差不是很大的,尽量合并,避免样品的分散; 对于一些量大的流分,可合可不合,反正量大,合与不合的效果是一样的(无非就是多占点空间); 从ODS上洗脱的流分,很可能是纯品;易结晶的样品,会在溶剂挥发的过程中结晶析出来,要注意观察;柱子被污染如何处理? 污染是正常的,见过那么多,也用过那么多ODS柱子,没有哪一根能在使用一段时间后保持“洁白无瑕”的。 被污染,一般情况下就是用甲醇冲洗,一直到冲洗液蒸干没有杂质为止,就可以进行下一批样品的分离了。 也许有人问:你这么处理彻底吗?回答是否定的,但是,对于ODS柱色谱,这样的处理已经足够了。 道理很简单,用ODS进行分离的样品,一般极性不会很小,70%甲醇/水基本能全部冲洗下来。现在已经用甲醇冲洗得差不多,在你对下一批样品进行分离的时候,上一批残留的杂质也就只有100%的甲醇能洗脱下来,根本不会影响。 如果一定要处理,可以采用甲醇:异丙醇=1:1冲洗,这种做法,算是终极做法了。
[color=#444444]用液相跑某一个样品,发现杂质峰比较多,欲将主峰物质进一步分离纯化,收集相应峰馏分。用普通的分析检测型的HPLC可以做到吗?还是用制备型色谱?只要收集到一点点就可以,然后可以做定性分析...我查文献里说都是用制备型色谱分离,希望懂色谱的高手指导一下,多谢[/color]
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]换了纯化器以后谱线很乱,很杂是什么原因







 我要推广仪器
我要推广仪器
 下载APP
下载APP