推荐厂家
暂无
暂无
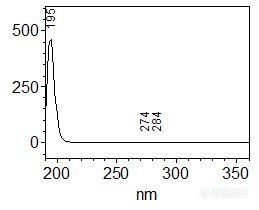
[align=center][b]左卡尼汀中间体及成品的分析——PDA及NQAD检测器对比[/b][/align]客户提供已知结构中间体2和中间体3,以及成品左卡尼汀单标样品,希望能够建立中间体液相分析检测方法。由中间体结构式可知其紫外吸收较弱,因此首先使用二极管阵列检测器——PDA,对其紫外吸收进行确认。紫外吸收光谱图如图1和图2所示,二者均为短波长吸收,最大吸收波长分别为195 nm和194 nm,最终选择的紫外检测波长为195 nm。[align=center][img=,259,210]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251523394585_463_2222981_3.jpg!w259x210.jpg[/img] [img=,252,204]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251523440193_5542_2222981_3.jpg!w252x204.jpg[/img][/align][align=center] 图1 中间体2紫外吸收光谱图 图2 中间体3紫外吸收光谱图[/align]接下来进行液相方法的建立。考虑到中间体的极性较强,故首先使用具有超高表面极性的反相柱CAPCELL PAK ADME,在100%水相条件下进行保留尝试,缓冲盐选择在低波长干扰较小的高氯酸钠体系。分析结果如图3所示,两中间体在反相系色谱柱上均无法得到保留,反相系色谱柱不适用于该项目分析。[align=center][img=,449,258]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251530523604_5470_2222981_3.jpg!w449x258.jpg[/img][/align][align=center]图3 CAPCELL PAK ADME色谱柱分析结果[/align][img=,451,170]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251530520833_9064_2222981_3.jpg!w451x170.jpg[/img]接下来,考虑利用阳离子交换模式和亲水性相互作用模式进行分析,以期得到良好保留和峰形。经过多方条件调整后,最终在亲水性相互作用色谱柱PC HILIC上得到良好保留结果,中间体分析谱图及放大图分别如图4-7所示。中间体在高氯酸钠体系下得到良好峰形,同时与死时间附近杂质峰取得了良好分离。应客户要求对其杂质进行了积分,积分表分别如表1-2所示(软件自动积分结果)。[align=center][img=,389,236]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251532088480_8928_2222981_3.jpg!w389x236.jpg[/img][/align][align=center]图4 中间体2分析结果[/align][align=center][img=,389,235]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251532091260_9820_2222981_3.jpg!w389x235.jpg[/img][/align][align=center]图5 中间体2分析结果放大图[/align][align=center] [/align][align=center]表1 中间体2分析结果积分表[/align][align=center][img=,624,541]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251532085430_4956_2222981_3.jpg!w624x541.jpg[/img][/align][align=center][/align][align=center][img=,509,298]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251536544585_463_2222981_3.jpg!w509x298.jpg[/img][/align][align=center]图6 中间体3分析结果[/align][align=center][img=,494,302]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251536551365_8367_2222981_3.jpg!w494x302.jpg[/img][/align][align=center]图7 中间体3分析结果放大图[/align][align=center] [/align][align=center]表2 中间体3分析结果积分表[/align][align=center][img=,562,566]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251537328944_2982_2222981_3.jpg!w562x566.jpg[/img][/align][align=center][/align][align=left][img=,549,237]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251538004262_3029_2222981_3.jpg!w549x237.jpg[/img][/align][align=left][/align][align=left]同时,我们也使用PC HILIC色谱柱进行了两中间体的共同分析,以期得到二者的基线分离结果,简化分析过程。然而在HILIC模式下,由于乙腈比例较高,且中间体自身需要在短波长下检测,因此可选的缓冲盐浓度及种类均有限;水相分别尝试使用0.1%磷酸溶液、20 mmol/L磷酸二氢钾溶液(磷酸调pH 2.5)、20 mmol/L磷酸二氢铵溶液(磷酸调pH 2.5)及不同浓度高氯酸钠溶液,结果均无法得到两中间体的分离;且在盐浓度不足时,中间体由于自身的季铵盐结构易产生吸附作用,很难得到良好峰形,典型谱图如图8所示。[/align][align=left][/align][align=center][img=,543,285]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251538324635_1675_2222981_3.jpg!w543x285.jpg[/img][/align][align=center]图8 0.1%磷酸条件PC HILIC分析结果[/align][align=center][/align][align=left]此外对柱温进行筛选,温度升高时保留时间有缩短趋势,但未见二者出现明显分离趋势,结果如图9所示。[/align][align=left][/align][align=center][img=,541,385]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251539139335_5664_2222981_3.jpg!w541x385.jpg[/img][/align][align=center]图9 不同柱温分析结果[/align][align=center][/align][align=left]考虑到中间体的整体紫外吸收较弱,应客户要求,使用高灵敏度气溶胶型检测器NQAD进行了分析对比。[/align][align=left]由于气溶胶型检测器NQAD要求流动相必须为挥发性盐,因此将水相中的高氯酸钠更换为50 mmol/L甲酸铵溶液(+0.1%甲酸)进行分析,中间体2、中间体3、左卡尼汀共同分析结果如图10-12所示。由于5 mg/mL样品浓度较大,在NQAD检测器上过载,出现信号平头峰或裂峰现象,把浓度稀释10倍后可得到正常峰形。但即使在大浓度样品分析中,未见死时间附近出现明显杂质峰,推测原因可能为杂质分子量较小,为挥发性杂质,无法在气溶胶型检测器上进行良好检出。此外,能够看到中间体在NQAD检测器上均出现多个明显色谱峰。[/align][align=left][/align][align=center][img=,674,407]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251540311232_3541_2222981_3.jpg!w674x407.jpg[/img][/align][align=center]图10 中间体2在NQAD检测器分析结果[/align][align=center][img=,670,402]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251550168795_4568_2222981_3.jpg!w670x402.jpg[/img][/align][align=center]图11 中间体3在NQAD检测器分析结果[/align][align=center][img=,677,395]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251550176905_730_2222981_3.jpg!w677x395.jpg[/img][/align][align=center]图12 左卡尼汀在NQAD上分析结果[/align][align=left]由于使用NQAD进行检测时,中间体均出现溶出多个色谱峰现象,怀疑为中间体的成盐离子导致,因此在小浓度下进样NaCl进行了排查,对比结果如图13所示。NaCl保留时间和较大杂质峰保留时间一致,可能为相应对离子。此外,在NQAD系统使用的甲酸铵流动相体系下,中间体2和3得到了分离。[/align][align=left][/align][align=center][img=,690,455]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251551127175_1278_2222981_3.jpg!w690x455.jpg[/img][/align][align=center]图13 不同样品NQAD比较图[/align][img=,637,239]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251551124165_6276_2222981_3.jpg!w637x239.jpg[/img][align=left][/align][align=left]综上所述,使用大阪曹達PC HILIC S5 4.6 mm i.d. × 250 mm色谱柱可完成左卡尼汀中间体及成品的保留,在紫外检测条件下获得较好峰形结果,能够与死时间附近的杂质峰取得良好分离。[/align]
制剂的中间物料如压片前的混合粉末,是叫中间体还是中间产品呢?原料药的中间产物呢?看有的资料写中间产品有的写中间体,区别大吗?
老师:今天我做了一个头孢中间体,分别用DMSO、D2O做溶剂,两张谱图相差很大,是溶剂效应吗?谱图是扫描的,效果不太好。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=49432]谱图及结构式[/url]





 400-628-5299
400-628-5299
 留言咨询
留言咨询
 400-876-8980
留言咨询
400-876-8980
留言咨询
 400-876-8980
留言咨询
400-876-8980
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


