如题:求助1,2,3-三唑(http://ng1.17img.cn/bbsfiles/images/2013/12/201312041240_480686_2834894_3.gif)的红外峰值,特征峰值
?氨气的红外光谱峰值位于3310-3350 cm^-1。?这个峰值是由于氨气分子中N-H键的伸缩振动引起的。具体来说,伯胺在3500 cm^-1处有吸收峰,仲胺在3400 cm^-1处有吸收峰,而叔胺则在3450 cm^-1处有吸收峰。这些吸收峰的位置和形状可以帮助区分不同类型的胺类化合物。? 此外,氨气的红外光谱特性还与其分子结构有关。氨气由一个氮原子和三个氢原子组成,形成金字塔形状的结构,这种非对称性使得氨气分子在振动或旋转时偶极矩会发生变化,从而在特定的红外波长上吸收光。特别是N-H键的伸缩和弯曲振动会导致偶极矩的变化,这在FTIR光谱中表现为明显的吸收峰。
[table=100%][tr][td]傅立叶红外光谱图~每个峰值都会对应一个特定的官能团吗?与是什么物质有关系吗?没关系的话,从哪可以找到官能团与峰值对应的图谱啊?谢谢谢谢~[/td][/tr][/table]
在做红外的时候,有的样本其实峰值应该差不多才对,实验过程中知识改变了他的一两个官能团,可是结果出来有的出现了迁移,而且还有一些不必要的杂峰,除了产品可能有杂质外,请问还有什么其他的原因?谢谢各位大神
请问专家,在分析XRD数据时,如何进行峰值的确定?峰值偏差对于分析的结果影响有多大?
红外光谱图的峰值用origin 8.0 如何添加
[table=100%][tr][td]在做样品光谱分析时,发现含有相同官能团的样品吸收峰峰值呈现出左移变大的规律,原因怎么分析呢?[/td][/tr][/table]
如何手工解谱从红外光谱图找波峰值
请教各位,有谁知道氧氮仪分析氮元素峰值强度为什么会返到零点以下的负值么?情况如图[img]http://ng1.17img.cn/bbsfiles/images/2017/05/201705221427_01_3234744_3.jpg[/img]
我的nicolet370型红外光谱仪,的干涉条文峰值很小,而且经常不扫描。请大家帮我分析仪下是什么原因。我将不胜感激。
正丁醇红外光谱分析急求!!每个官能团对应的峰值各是多少?
[b][color=#444444][url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]分析中,测量的方式是峰值吸收,所以吸光度反映其大小是对的吗?[/color][/b]
如何根据红外吸收光谱峰值判定Wavenumbers

[img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903071706_137216_1631661_3.jpg[/img]小弟接触红外时间不久,请教大家一个问题QQ:57536786E-mail:sunzhimems@163.com愿听赐教!图是我的薄膜样品,分别从薄膜的二个面入射而做出来的红外图。简单看来他们的峰数基本一致,但是在指纹区他们的峰值相差了大约一半啊?教科书上说:峰值与跃迁几率的大小和振动偶极距变化的大小有关。跃迁几率大和振动偶极距变化大则吸收峰强。那么对与我做的同一种材料的二个侧面,他们的峰值相差一半,应该怎么解释一下那?! 还是由于跃迁几率,偶极距变化吗? 二个侧面的成分含量不一样吗?!
偶没学过红外光谱相关的课程,但毕设中涉及到红外光谱的分析,在此特向大家求助,往XDJMs帮偶一下,先谢谢啦~请帮忙分析一下图中曲线(1)(2)(3)(4)官能团的变化 红外光谱图及峰值如下:(1)的峰值分别是3431cm,2974cm,1296cm,1049cm,423cm。(2)的峰值分别是3436cm,2925cm,1296cm,867cm,627cm(凸)。(3)的峰值分别是3431cm,2973cm,1292cm,867cm,628cm(凸)。(4)的峰值分别是3437cm,2958cm,1297cm,868cm,623cm(凸)。
请教高手,做红外,空白用KBr压片,显示的图谱按“计算”的按钮,会自动检出峰值标示在图谱上,然后接着扫描样品片,出现图谱之后按“计算”的按钮,没有峰值标示在图谱上???为什么呢|???
[color=#444444]采用原位红外分析随着温度的升高,NH3在分子筛表面的脱附情况,随着温度升高,一方面由于脱附导致峰值减小,另一方面是不是由于温度的升高导致峰值有所增加,求问进行半定量分析时,如何消除温度的影响[/color]
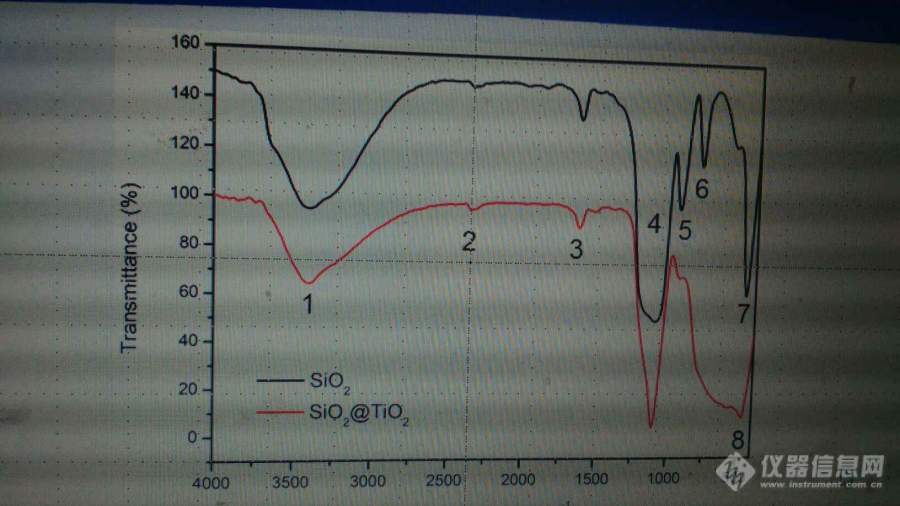
[color=#444444]请求各位告知1~8是什么物质的峰值,谢谢。[/color][color=#444444][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131404588767_8288_1806906_3.jpg!w690x387.jpg[/img][/color]
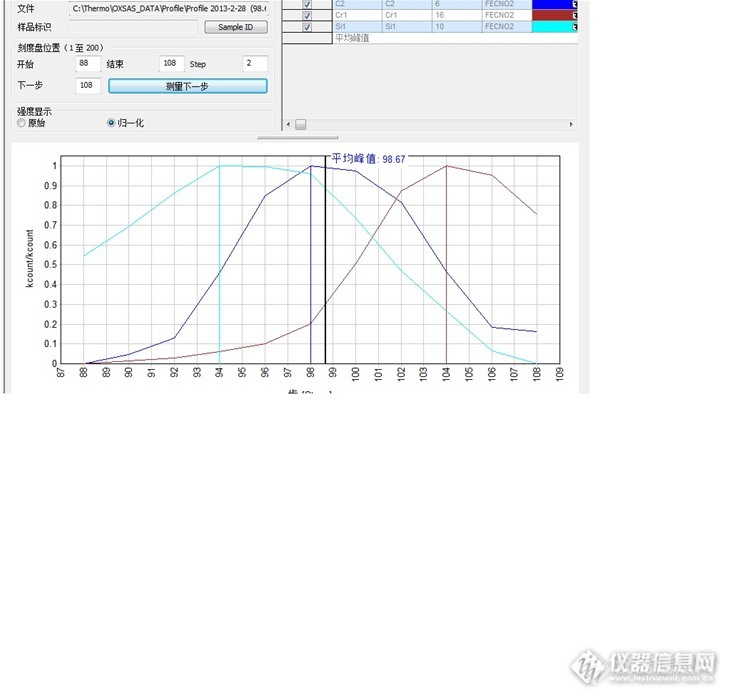
今天按照维护计划更换了真空泵油,按照维护手册的方法,更换真空泵油后要对设备进行积分描迹。但是描迹完后看峰值有些疑惑。见图:1、设备刚买后进行的描迹,时间在2011年4月http://ng1.17img.cn/bbsfiles/images/2013/02/201302282108_427682_1617441_3.jpg2今晚刚完成的描迹http://ng1.17img.cn/bbsfiles/images/2013/02/201302282110_427684_1617441_3.jpg看图后,谁能给解释下为什么现在的几个峰值相差距离很大啊,而当时几乎都在一块的,都是98的。相差间距大对设备描迹取峰值有影响吗,能否代表现在的是光通过最大的位置。是否需要重新选取几个接近的代表通道呢?今天早上重新选择标样进行描迹,几个峰值以在一个点上,确定是原来标样选择错误,希望大家吸取教训,不要犯同类型错误了。
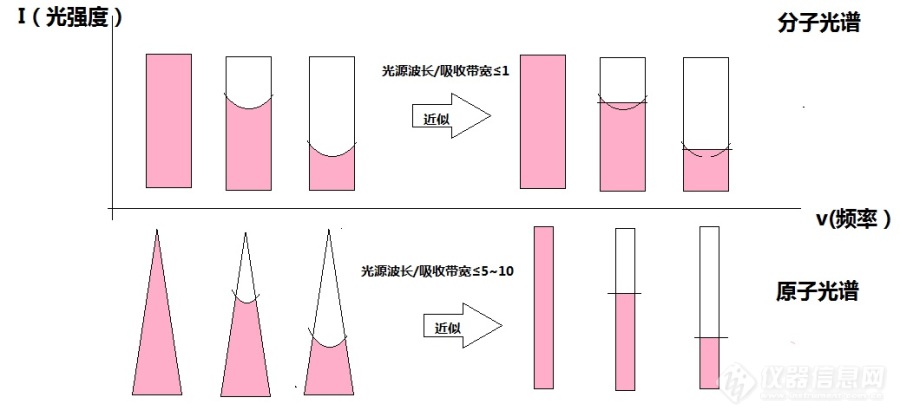
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]中经常提到,因为空心阴极灯的光谱很窄(0.002nm),计算积分吸收系数不易,故以峰值吸收值代替积分吸收。但实际根据Lambert-Beer定律,实际测定的是光的强度I0和I,这本身就是积分吸收值啊,得到的a相当于平均吸收系数,和峰值吸收有什么关系呢?[img=,690,320]http://ng1.17img.cn/bbsfiles/images/2018/05/201805162237123439_3468_1340227_3.jpg!w690x320.jpg[/img]
我所采用的是FID,毛细管柱,在做分析时,发现主峰峰值偏大,其它峰值偏小,这是为什么?该如何解决?
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法的基本原理中的积分吸收和峰值吸收还是不太明白积分吸收稍微有点明白了,但是峰值吸收又是什么意思呢?峰值吸收为啥要用锐线光源呢?最终实际应用的时候,测定的是吸光度,吸光度和单位体积内被测元素基态原子数有关系的,这又和峰值吸收没关系了吧?哪位大侠能给小弟详细解释一下
在给一个学生侧样品时,他样品袋子上写的AlN 但是测得的峰值有二三十万 用软件查了一下也没查出是什么东西来 请问什么东西的峰值有这么高啊 [em0904]
求教各位版友~对这台机器还是比较菜,求大家帮忙~~着急~~GCMS 6890-5973,做测量审核样品——大米粉中的毒死蜱含量测定使用内标法定量,在制作标准曲线时,各点的内标含量均为0.5ppm(毒死蜱标准品标明用丙酮稀释,内标为之前储存也,接手前就一直使用丙酮稀释)样品按照国标处理,加内标亦为0.5ppm,最终用正己烷定容1mL标准曲线点跑出的谱图峰值很低,内标峰值为500-,而样品所得谱图内标峰值为1100+怕是基质问题,做过如下标准品①将购买的毒死蜱标准品用丙酮稀释,加内标,用正己烷定容1mL②将购买的毒死蜱标准品用丙酮稀释,加内标,用丙酮定容1mL结果相同~由于大米粉中毒死蜱含量未知所以只能确定内标的峰值过低,现在谱图不方便放上来,晚些时候来放图,期限又要到了,着急T.T求大家帮忙~~谢谢!!~万分感激!!~========================================================================2013.11.13 关于此次出现问题的最终解决方法反馈:2013.11.08检测结果已经寄送出去,11.11被对方签收,目前结果还没有出来,出后会补发的~现只总结下该问题的最终解决办法~~虽然具体原因不清楚,不过希望写出来能够有点帮助~~测过几次都出现同样问题后,另一位同事根据她测苏丹红的经验,猜测是不是应该把配置好的标准品也过下柱子于是将标准品配置好后过柱子备用,同时防止再出现问题,更换了新的衬管标准品与样品上机后,两者内标峰值基本保持一个水平了~约为30,000+标准曲线R值较好,回收率试验三次,平均回收率为104%以上就是基本的情况哦~~换衬管后也有侧过没过柱子的标准品,与换衬管前相似所以,为什么过了柱子后峰值变大,原因不明不知大家有何高见~~
TVOC标准曲线具体怎么做?反吹多久?热解析多久才好?我做出来的锋特别小,用的东西分析的仪器,分流和尾吹应该怎么调好呢?直接进样,溶剂峰特别大,注射到采样管里,峰值又特别小了
实现峰值吸收的两个重要条件是?空心阴极灯为什么满足着两个条件?
各位老师,我现在在学习原子光谱,有点疑惑请大家不吝赐教。原子发射的光谱是锐线光源,测定积分吸收能做到么?是不是和[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]一样测定的是峰值发射值。其次,原子发射光谱中既有原子化的,又有离子化的,原子化和离子化的比例是恒定的么,如果不恒定,那定量分析不是有困难?还是需要加内标才行?
普析PF32 载流:5%硝酸 ,硼氢化钾现用限配的最近做砷的,这几次做的时候,荧光值变小的很明显。浓度10ug/l 的时候最高点只有200的荧光值,载流空白只有10左右 。之前每天做原子荧光的时候,峰值每次都比前一次小。换了蠕动泵泵管后结果影响不大。汞灯的荧光值也变小了,但没有As那么夸张。进样时观察灯丝是点燃的,样品也明显变少。现在也不知道是什么原因。
[em16] 我们正在使用一台美国LECO 344红外碳硫分析仪,本人才疏学浅,愣是搞不清楚何为"分析用谱线",是所谓C.S的积分曲线(根据电脑输出绘制的峰值波形图)还是C.S的标准工作曲线(用不同C.S含量的标样分析绘制的曲线),请各位高手赐教!
[color=#444444]同一种物质A-CPD在做红外光谱分析的过程中,不同的人测得的峰值数据略有差异,这是什么原因?两人都对还是其中一个人是对的?[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2019/0218/bw121h7178788_1550477058_680.jpg[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2019/0218/w120h7178788_1550477059_669.jpg[/img][/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP