推荐厂家
暂无
暂无
7月23日,国家药典委公示了17个中药标准草案:冠心丹参滴丸、消食退热糖浆、金嗓开音颗粒、金嗓利咽丸、葶苈子、松花粉、芥子、巴豆、紫苏子、栀子、王不留行、水红花子、诃子、咳特灵片、板蓝根含片、复方塞隆胶囊、复方丹参滴丸涉及的9个药材和饮片品种主要修订内容如下:葶苈子:修订葶苈子药材、葶苈子饮片、炒葶苈子饮片的性状项及显微鉴别项。修订葶苈子药材、葶苈子饮片和炒葶苈子饮片的薄层色谱鉴别项。松花粉:增订了松花粉浸出物项。芥 子:修订芥子药材薄层色谱鉴别项、芥子饮片和炒芥子饮片的薄层色谱鉴别项。巴 豆:增订生巴豆饮片的检查、含量测定项。紫苏子:修订炒紫苏子水分检查项。栀 子:修订栀子来源项采收时间、性状项、规范对照品名称。王不留行:增订炒王不留行饮片总灰分检查项。水红花子:修订了水红花子的薄层色谱鉴别项。诃 子:增订诃子饮片鉴别、检查项、浸出物项;增订诃子肉饮片鉴别项、检查项、浸出物项。
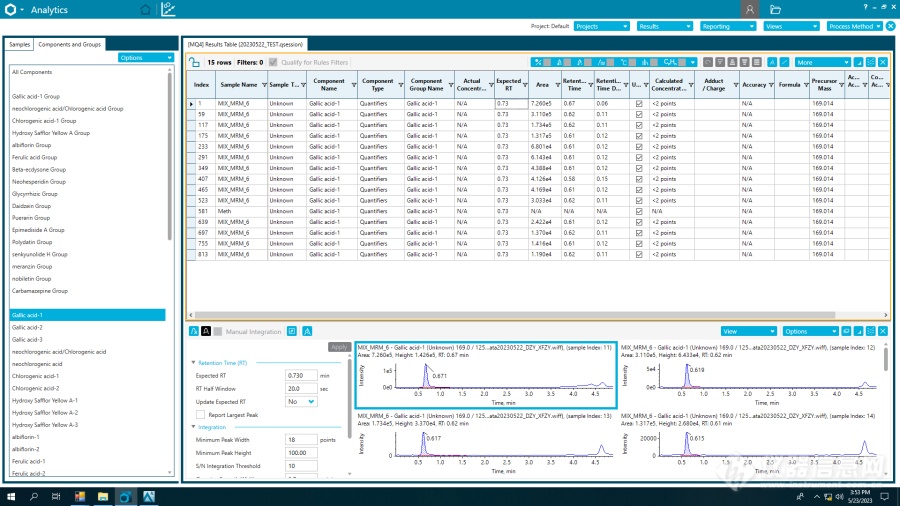
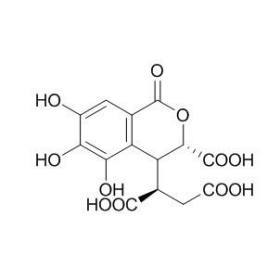
建了[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析方法,想同时检测没食子酸、新绿原酸和绿原酸和其他几种成分,如羟基红花黄色素A、阿魏酸等,但对照品建立方法时连续进样6针,没食子酸、新绿原酸和绿原酸的峰面积逐渐降低,怀疑是降解,但拿到HPLC上检测是没有问题的, 峰面积与新配制的相同。请问是什么原因导致的,而且对对照品为混标,其他成分都没有降解的情况,很稳定,可以排除进样的原因,如图[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2023/05/202305281454099518_1703_3962371_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2023/05/202305281454257768_9259_3962371_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2023/05/202305281454341112_6631_3962371_3.png!w690x387.jpg[/img]
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo






 留言咨询
留言咨询
 留言咨询
留言咨询

 400-860-5168转2499
留言咨询
400-860-5168转2499
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP