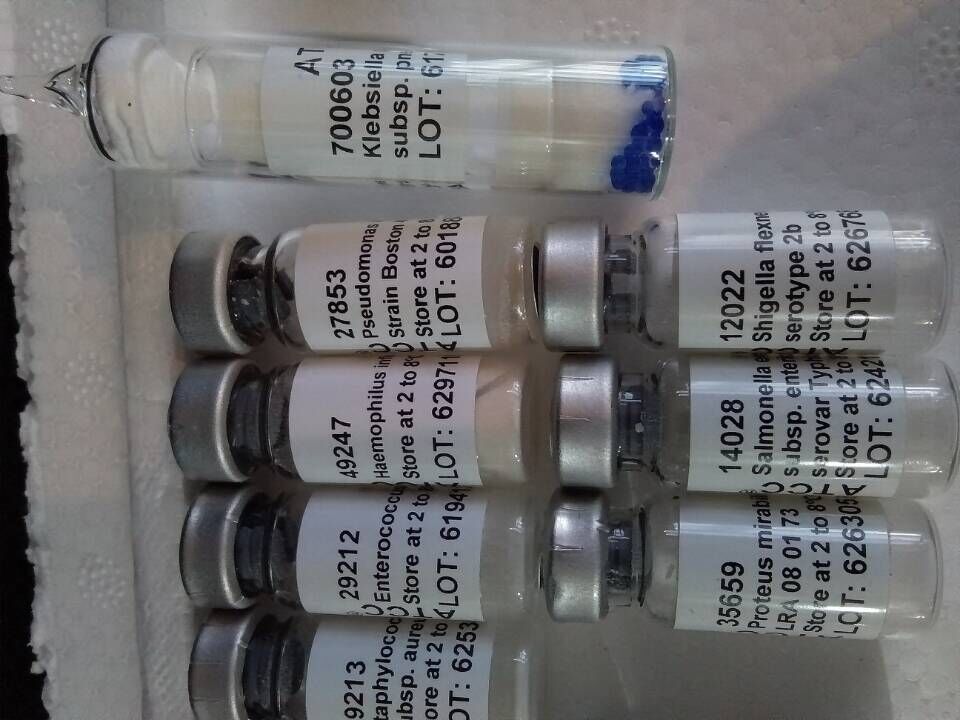
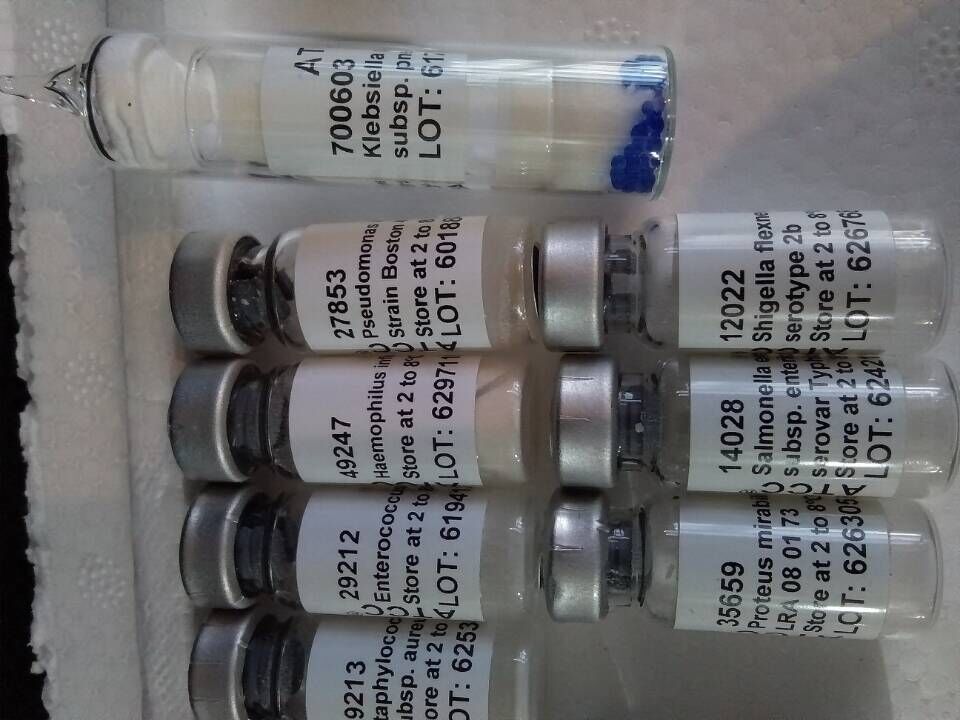
推荐厂家
暂无
暂无

在外聚餐时,餐桌上常会有赠送的餐前小吃——花生米,不过,如果你在金灿灿的花生米中无意夹到一颗变黑的,这时可千万不要往嘴里送,因为它们有可能已经被黄曲霉毒素污染而可能会对人体产生一定的危害。那什么是黄曲霉毒素呢?http://ng1.17img.cn/bbsfiles/images/2015/07/201507071036_553899_2984502_3.jpg1黄曲霉毒素 黄曲霉毒素是一种由黄曲霉和寄生曲霉等真菌经过聚酮途径产生的次生代谢产物,是一组结构类似的化合物总称。迄今为止,已发现的黄曲霉毒素至少包含有黄曲霉毒素B1、B2、G1、G2、M1、M2、P1、Q1、H1、GM、B2a、G2a及毒醇等20种左右结构相似化合物。 通常黄曲霉毒素存在于土壤、动植物、各种坚果特别是花生和核桃中。在大豆、稻谷、玉米、通心粉、调味品、牛奶及奶制品、食用油等制品中也可能会发现黄曲霉毒素。2黄曲霉毒素在食物中的限量标准 根据我国国家标准GB2761-2011《食品中真菌毒素限量》的规定,我国食品中黄曲霉毒素B1允许量标准为:玉米及其制品、花生及其制品中不得超过20μg/kg;稻谷、糙米、大米和其他植物油脂中不得超过10μg/kg;小麦、大麦、其他谷物、发酵豆制品和其他熟制坚果及籽类中不得超过5μg/kg;部分调味品(如酱油、醋、酿造酱)中不得超过5μg/kg;婴幼儿配方食品中不得超过0.5μg/kg。乳及乳制品和特殊膳食用食品中黄曲霉毒素M1限量不得超过0.5μg/kg。3黄曲霉毒素的检测方法(1)薄层色谱法(TLC法) TLC法是测定黄曲霉毒素的经典方法,是我国测定食品及饲料中黄曲霉毒素B1的标准方法之一(GB/T5009.23-2006)。适用于各种食品中黄曲霉毒素B1、B2、G1、G2的测定。(2) 免疫分析法 免疫化学分析法是以抗原抗体的免疫化学反应为基础进行抗原抗体含量测定的方法。用于黄曲霉毒素测定的免疫分析法主要有免疫亲和层析净化—荧光光度法、免疫亲和层析净化—高效液相色谱法、酶联免疫吸附法等。 免疫亲和层析净化—荧光光度法和免疫亲和层析净化—高效液相色谱法 免疫亲和层析技术的方法特异性好、灵敏度高,是目前我国现行国家标准推荐的方法之一(GB/T18979-2003),适用于玉米、花生及其制品(花生酱、花生仁、花生米)、大米、小麦、植物油脂、酱油、食醋等食品中黄曲霉毒素的测定。 酶联免疫吸附法(ELISA) ELISA法是应用抗原抗体特异性反应和酶的高效催化作用来测定黄曲霉毒素含量的免疫分析方法,其相对应的标准是GB/T5009.22-2003(第二法),适用于粮食、花生及其制品、薯类、豆类、发酵食品及酒类等各种食品中黄曲霉毒素B1的测定。4黄曲霉毒素的预防与控制措施(1) 挑选霉粒法 因黄曲霉毒素主要集中在霉坏、破损、皱皮、变色和虫蛀等的粮粒中,这些带毒颗粒比健康颗粒轻,外表也较易辨认,可用机械或人工掏除。(2) 植物油加碱去毒法 油料种子受黄曲霉毒素污染后,榨出的油中含毒素,可用碱炼法去毒。因为不溶于水的黄曲霉毒素在碱性条件下可形成香豆素钠盐而溶于水,故加减后用水洗可将毒素去除。(3) 加水搓洗法 在淘洗大米时,用手搓洗,随水倾去悬浮物,如此反复5~6次,煮熟后可去除大部分毒素。(4) 吸附去毒法 植物油受黄曲霉毒素污染,可利用活性白土和活性炭吸附,效果较好。(5)高温高压去毒法 黄曲霉毒素较耐高温,在280℃高温下才能分解,因此一般烹调温度下难以消除。但高温高压下去毒效果较好。(6) 碾磨去毒法 在粮食中,黄曲霉毒素大部分集中于脂肪较多的胚体和糠皮等部位。稻谷经精碾后95%的毒素可去除。玉米磨粉也有类似的效果。
请教一下各位:酿酒中所用的米曲霉,是属于食品添加剂还是辅料?成品的标签中需要标示吗?
一、概述赭曲霉毒素又称棕曲霉毒素,是曲霉属和青霉属的一些菌种产生的一组结构类似,主要危及人和动物肾脏的有毒代谢产物,分为赭曲霉毒素A、赭曲霉毒素B、赭曲霉毒素C、赭曲霉毒素D四种化合物,其中赭曲霉毒素A(OA)分布最广、产毒量最高、毒作用最大、农作物污染最严重、与人类关系最密切,是一种强力的肝脏和肾脏毒素。OA是异香豆素联结L一苯丙氨酸在分子结构上类似的一组无色结晶化合物,溶于极性溶剂和稀的碳酸氢钠水溶液中,微溶于水。在紫外线下OA呈绿色荧光。该化合物相当稳定,在乙醇中置冰箱避光可保存一年。世界各国均有从粮食中检出oA的报道,但其污染分布很不均匀,污染的谷物主要为麦类及玉米,产毒适宜温度为20~30℃。二、检测方法通常采用薄层色谱法和高效液相色谱法测定。薄层色谱法原理1.原理用三氯甲烷-0.1mol/L磷酸或石油醚一甲醇/水提取样品中的OA,样品提取液经液一液分配后,根据其在365nm紫外线灯下产生黄绿色荧光,在薄层板上与标准比较测定含量。2.试剂石油醚(沸程为60~90℃或30~60℃)、甲醇、三氯甲烷、甲苯、乙酸乙酯、甲酸、冰乙酸、乙醚、苯一乙腈(98+2)、0.1mol/L磷酸溶液(称取11.5g 85%的磷酸加水稀释至1000mL)、2mol/L盐酸溶液(量取20mI。盐酸,加水稀释至120mL)、4%氯化钠溶液、0.1mol/L碳酸氢钠溶液(称取8.4g碳酸氢钠,加适量水溶解,并用水稀释至1000mL)、硅胶G。OA标准储备溶液:用苯一冰乙酸(99+1)配成40pg/mL OA储备溶液,并用紫外分光光度计测定其浓度。置冰箱中避光保存。OA标准使用液:精密吸取储备溶液,用苯稀释成OA标准工作液(0.5μg/mL),置冰箱中避光保存。3.仪器与设备小型粉碎机,电动振荡器,玻璃板(5cmx 20cm),薄层涂布器.展开槽(内径25cm、宽6crn、高4cm),紫外线灯(365nm),微量注射器(10μL、50μL),具0.2mL一尾管的10mL浓缩瓶。所有玻璃仪器均需用稀盐酸浸泡,用自来水、蒸馏水冲洗。4.分析步骤(1)提取称取20g样品,精确至0.01g,置于200mL具塞锥形瓶中,加入100mL一三氯甲烷和10mL 0.1mol/L磷酸溶液,振荡30rnin后通过快速定性滤纸过滤。取20mL滤液置于250rnL分液漏斗中,加50mL 0.1mol/L碳酸氢钠溶液振摇2min,静置分层后,将三氯甲烷层放入另一个100mL分液漏斗中(少量乳化层,或即使三氯甲烷层全部乳化都可放入分液漏斗中)。加入50mL 0.1mol/L碳酸氢钠溶液重复提取三氯甲烷层,静置分层后,弃去三氯甲烷层(如果三氯甲烷层仍乳化,弃去,不影响结果)。碳酸氢钠水层并入第一个分液漏斗中,加约5.5mL 2mol/L盐酸溶液调节pH2~3(用pH试纸测试),加入25mL三氯甲烷振摇2min,静置分层后,放三氯甲烷层于另一盛有100mL水的250mL分液漏斗中,酸水层再用10mL三氯甲烷振摇提取、静置。将三氯甲烷层并入同一分液漏斗中。振摇、静置分层,用脱脂棉擦干分液漏斗下端,放三氯甲烷层于一75mL蒸发皿中,将蒸发皿置蒸汽浴上挥干。用约8mL一三氯甲烷分次将蒸发皿中的残渣溶解,转入具尾管的10mL浓缩瓶中,置80℃水浴锅上用蒸汽加热吹氮气浓缩至干,加入0.2mL苯一乙腈溶解残渣,摇匀,供薄层色谱点样用。(2)测定取两块薄层板,在距薄层板下端2.5cm的基线上用微量注射器滴加两个点:在距板左边缘1.7cm处滴加OA标准溶液8μL(浓度0.5μg/mL),在距板左边缘2.5cm处滴加样液25μL,然后在第二块板的样液点上加滴OA标准溶液8μL(浓度0.5μg/mL)。点样时,需边滴加边用电吹风吹干,交替使用冷热风。(3)展开在展开槽内倒人l0mL乙醚或乙醚一甲醇一水(94+5+1),先将薄层板纵展至离原点2~3cm,取出通风挥发溶剂1~2min后,再将该薄层板靠标准点的长边置于同一展开槽内的溶剂中横展,如横展剂不够,可添加适量,展至板端过1min,取出通风挥发溶剂2~3min。在另一展开槽内倒入10mL甲苯一乙酸乙酯一甲酸一水(6+3+1.2+0.06)或甲苯一乙酸乙酯一甲酸(6+3+1.4),将经横展后的薄层板纵展至前沿距原点13~15cm,取出通风挥干至板面无酸味(约5~10min)。(4)观察与评定 将薄层色谱板置365nm波长紫外线灯下观察,两板比较,若第二块板的样液点在OA标准点的相应处出现最低检出量;而第一板相同位置上未出现荧光点,则样品中的OA含量在本测定方法的最低检测量10μg/kg以下;如果第一板样液点在与第二板样液点相同位置上出现荧光点,则看第二板样液的荧光点是否与滴加的标准荧光点重叠,再进行以下的定量与确证试验。(5)稀释定量 比较样液中OA与标准OA点的荧光强度,估计稀释倍数。经稀释后测定含量时,可在样液点的左边基线上滴加两个标准点,比较样液与两个标准OA荧光点的荧光强度,半定量。(6)确证试验用碳酸氢钠一乙醇溶液(在100mL水中溶解6.0g碳酸氢钠,加20mL乙醇)喷洒色谱板,在室温下干燥,于长波紫外线灯下观察,这时OA荧光点应由黄绿色变为蓝色,而且荧光强度有所增加,再估计样品中OA。





 400-860-5168转4139
400-860-5168转4139
 留言咨询
留言咨询
 400-860-5168转6008
留言咨询
400-860-5168转6008
留言咨询
 400-860-5168转1522
留言咨询
400-860-5168转1522
留言咨询








 我要推广仪器
我要推广仪器
 下载APP
下载APP