推荐厂家
暂无
暂无
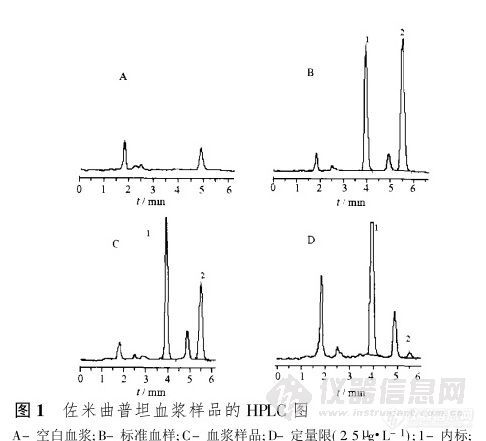
【作者】 蔡佳; 蒋新国; 陈钧; 熊志刚; 金樑; 【Author】 CAI Jia,JIANG Xin-guo~*,CHEN Jun,XIONG Zhi-gang,JIN Liang(Department of Pharmaceutics,School of Pharmacy,Fudan University,Shanghai 200032,China) 【机构】 复旦大学药学院药剂学教研室; 复旦大学药学院药剂学教研室 上海200032; 上海200032; 【摘要】 目的建立大鼠血浆中佐米曲普坦的高效液相测定方法,并研究大鼠不同途径给药后的药动学。方法采用甲基叔丁基醚为溶剂,提取药物。以0.05%三乙胺(用磷酸调至pH 2.70)-乙腈(92∶8)为流动相,色谱柱为Dikma Diamonsil C18柱(4.6 mm×200 mm,5μm),流速1.2 mL.min-1,荧光检测的激发波长225 nm,发射波长360 nm。结果佐米曲普坦在2.5~1 000μg.L-1内线性关系良好(r=0.999 7)。高、中、低3种浓度的提取回收率分别为90.10%,91.75%,86.79%,方法回收率分别为103.55%,94.49%,98.79%,日内和日间RSD均小于4%,最低检测限为1μg.L-1。计算出灌胃、静注、鼻腔给药途径主要药动学参数分别为:t1/2(2.03±0.88)h,ρmax(144±28)μg.L-1,tmax(0.85±0.14)h,AUC0~t(442±110)μg.h.L-1;t1/2(1.40±0.12)h,ρmax(567±55)μg.L-1,AUC0~t(1 075±128)μg.h.L-1;t1/2(1.48±0.23)h,ρmax(304±34)μg.L-1,tmax(0.65±0.14)h,AUC0~t(685±43)μg.h.L-1。结论该方法操作简单、快速、准确、重现性好,适用于大鼠血浆中佐米曲普坦浓度的检测及其药动学研究。 【关键词】 佐米曲普坦; 高效液相色谱法; 药动学;http://ng1.17img.cn/bbsfiles/images/2012/09/201209022115_388005_1838299_3.jpg
各位大神,我用DNS分光法测糖蜜中还原糖,网上没买到相对应的质控样,只买到了95%的D-葡萄糖标准品,我打算用分析纯无水葡萄糖做标准曲线,用95%的D-葡萄糖标准品做准确度验证,因为样品的还原糖高达48%,不打算做加标回收率,如何把95%的D葡萄糖标准品配制成10mg/ml的标准溶液做准确度验证?

在分析技术的园地里发现许多朋友提到有关标准品的各种问题。其实我想主要的就是很少有人把这个题目讲解的透彻一点,让大家都能够一次彻底了解。自己在美国工作了30余年,也一直在分析的领域上,所以决定花时间把它写下来与大家共享。也算是抛砖引玉,能够给大家一个思考的机会,同时大家也互相交流,相得益彰。闲话少说,言归正传。分析的方法分成两种,一种就是绝对的方法,是不需要标准品,当然使用这种方法必须使用相当纯度的化合物。分析的方法譬如元素分析,DSC,NMR, 滴定,电化学等等,这些方法都可以用来决定化合物的纯度。如何取得相当纯度的化合物,一般我们就用色谱,首先肯定色谱的纯度(其实是均匀度homogeneity),当然在注射相当量的前提下(要能察觉到0.1%的杂质),只能看见一个主峰,也就是说均匀度接近100%(根据峰面积计算)。此时我们拿这个接近100%均匀度的化合物来定性,我们必须确认其专有性(specificity,属性)的存在,然后再定量得出此化合物的纯度。这里说的纯度是指化学组成分而言(composition)。因为这些方法没有办法测出金属盐类,或非金属盐类。因此我们要有另外的方法来决定离子的存在与其含量。当然还有其它成分如水分,有机溶剂等等。当我们把所有的成分定出来之后,总和就是我们这个化合物的组成成分。有了这些数据,自然我们可以求得物质平衡。所以,任何标准品的订定都必须经过这个程序。另外一种分析的方法就是相对的方法。顾名思义,就是需要标准品来对比。除了上述的绝对的方法外,其它的方法都是相对的。看看我们做色谱,就是一个最好的例子。我们需要标准品来定性及定量我们所要分析的样品。我以前曾经开发过新药(New Molecular Entity),标准品就需要自己来定了。经过色谱的分离,纯化干燥后,进行了如上段所叙述的定性,定量工作,我们取得了化学纯度。这个标准品,我们把它叫做原始标准品(primaryreference standard)。如果说化学纯度是95%,也就是说每次我秤了1毫克,里面含有此化合物0.95毫克。因为这种原始标准品,制备很繁杂而且获得的量也不大(在一定时间内),所以没有必要每次都使用这种原始标准品来进行色谱或其它相对分析方法分析样品。因此我们可以拿我们的化学原料药(一般就是工艺开发后的产品)来订为二级标准品(secondaryreference standard)。有人把它说成工作标准品,其实是有商酌余地的。工作标准品也可以是原始标准品,我们不需费力,可以采购而来。我们所要的做的工作,就是把这个二级标准品做色谱分析,然后用原始标准品来做工作标准品,完成定性,定量。如此,我们可以根据峰面积,及原始标准品的相应比(responsefactor = peak area / concentration)求出二级标准品的含量。以后我们做色谱的时候,就可以把这个订好的二级标准品拿来做我们的工作标准品来分析我们的样品。前面提到原始标准品的由来。可以自己定,当然也可以购买。在美国就是美国药典(USP),它不是一个官方机构。但是只要有官方承认的标准品,自然就去买,不管贵贱。如果太贵,那就买一次,然后用来做原始标准品,定出我们自己的二级标准品。一般美国药典的标准品都有规定干燥的程序。在执行干燥完成后,如果没有表明纯度,那就是100%。当下一个批次原始标准品发行的时候,前一批次自动失效。也就是有效期完全看新的批次何时出现。这样也避免了稳定性的考察。当然我们可以拿这个原始标准,来考察我们二级标准品的稳定性。国内的情形我不太清楚,我相信只要官方机构认可的标准品,都可以直接使用的。一般最常用的化学原料药的美国药典标准品,200毫克在200美元左右。如果我们使用微量天平,每次用量1.000毫克,那就可以每次使用原始标准品来做色谱,就避免再去设定二级标准品的麻烦了。另外我想大家都知道,当我们定量的时候,同一个标准品,我们必须秤两次,配置两个不同浓度的工作液。这样可以确认两个工作液是否配置正确。关于到底应该进样几针。我一向的做法就是第一个进样六针,做为系统适应度(system suitability)的检查,其它的任何样品,一律三针。有的人说两针就可以了。我要提醒的是两点决定一条线,得出的统计结论那是数学公式计算出来的。三点才能决定一个面,也就是高斯的分配图(GaussianDistribution)。所以至少在统计学上三点比两点得出的结果要好。又有人说了,如果我一个样品,要走60分钟,那不是完蛋了。首先,是否有必要要走60分钟。我认为最理想的方法是在30-45分钟(当然看功力了),这是指对杂质的分析,必须走梯度。我曾经用5厘米的柱子,梯度,30分钟分出17个化合物,包刮两个主要成分,两个抗氧剂,其它的就是杂质,分解物。如果走等梯度,一般是不超过10分钟为原则。这样就没有问题了。如果真的要走60分钟,那就必须选择好的仪器。并不是所有HPLC的仪器都可以无忧的走完全部样品。当然,柱子也十分重要。扯远了,以后有时间再分开来讨论。







 400-628-5299
400-628-5299
 留言咨询
留言咨询
 400-827-8618
留言咨询
400-827-8618
留言咨询
 400-860-5168转0843
留言咨询
400-860-5168转0843
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP