推荐厂家
暂无
暂无
如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
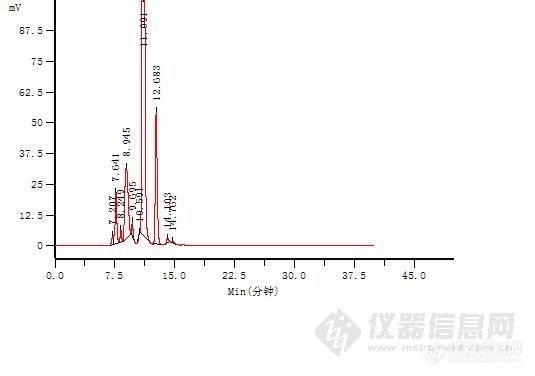
高效分子排阻色谱法测定注射用盐酸头孢替安高分子杂质头孢替安是杀菌性头孢菌素类广谱抗生素,头孢替安不但对革兰氏阳性菌有效,而且对革兰氏阴性菌。如流感嗜血杆菌,大肠杆菌、克雷白氏菌、奇异变形杆菌等的作用更强。对肠杆菌,枸橼酸杆菌、吲哚阳性变形杆菌等,也有抗菌作用头孢替安在肺中药物浓度较高,其它脏器和肌肉也有一定的浓度。临床应用于敏感菌所导致的感染,如肺炎、支气管炎、胆道感染、腹膜炎、尿路感染以及手术后或外伤引起的感染和败血症等。其基本结构同已上市的的头孢菌素类抗生素一样,头孢替安也会形成高分子聚合物,也会在临床使用中引发速发型过敏反应。对患者危害极大。已有的注射用盐酸头孢替安国家药品标准未将盐酸头孢替安高分子聚合物列为检定项目,国内的药学研究也未见头孢替安高分子聚合物的研究和报道。从临床用药安全性考虑,根据中国药典2010年版二部附录凝胶色谱原理。采用常用的葡聚糖凝胶G-10检测聚合物时由于头孢替安分子结构自身的原因,头孢替安不能完全缔合,因些我们采用高效分子排阻色谱法,以球状蛋白色谱用亲水硅胶为填充剂 TOSOH TSKgelG2000SW(7.5*300mm),测定注射用盐酸头孢替安高分子杂质1.仪器与试剂(1)仪器:岛津LC-10ATvp泵 岛津SPD-10AVP紫外可见光多波长检测器 浙大2010色谱数据工作站 色谱柱:TOSOH TSKgelG2000SW(7.5*300mm) (2)试剂: 乙腈 (色谱纯,天津市四友生物医学技术有限公司) 磷酸氢二钠(分析纯,北京化学试剂公司) 磷酸二氢钠(分析纯,北京化学试剂公司)双蒸水 (自制)2 色谱条件色谱柱:TOSOH TSKgelG2000SW(7.5*300mm)流动相:磷酸盐缓冲液(p H:6.8[/color
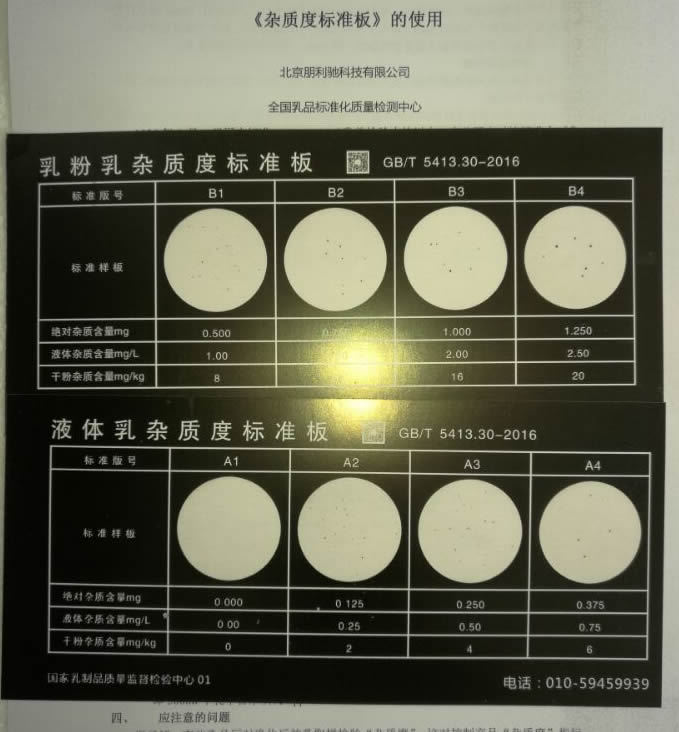
副产盐酸中含有大量的杂质成分,我估计了一下,肯能含有硫酸根,乙酸,氯乙酸,二氯乙酸等成分,硫酸根可以用氯化钡定量,但其他杂质怎么测定呢?有比色法或滴定法可以进行检验的吗?请各位大侠帮忙解决一下,谢谢!





 400-860-5168转1610
400-860-5168转1610
 留言咨询
留言咨询
 400-860-5168转1610
留言咨询
400-860-5168转1610
留言咨询
 400-860-5168转1610
留言咨询
400-860-5168转1610
留言咨询









 我要推广仪器
我要推广仪器
 下载APP
下载APP