有时候一个检测项目,要检测几种甚至几十种物质,以前如果用单一标准品的话,是不是需要把相应的标准品都备齐呢?那样的花费有点贵了,如果用混标的话,一次性就搞定了,成本是不是要降低很多呢?
发现现在我们的很多消费品的标准都是美国的,中国的消费品的标准有哪些?国标,企业标准,行业标准,有几许?欢迎喜欢收藏的朋友能分享下,感谢!

标准物质正十六烷,含量是100ng/微升,用来检测GC的稳定性先是采用程序升温,从50升至160度,速率为10度/分钟,载气流速1.5,分流比10:1,得出的图谱如下图所示,出现很多杂峰,未知哪个是标准物质。http://ng1.17img.cn/bbsfiles/images/2012/09/201209070908_389156_2210929_3.jpg然后改条件,不进行程序升温,直接就把温度保持在160度,分流比设为20:1,载气流速改为1.0,此时图谱如下图所示,峰分开了,但是不止一个,http://ng1.17img.cn/bbsfiles/images/2012/09/201209070909_389158_2210929_3.jpg再改条件,载气流速改为1.5,得图如下,http://ng1.17img.cn/bbsfiles/images/2012/09/201209070909_389159_2210929_3.jpg疑问:标准物质应该是很纯的,除了溶剂峰就是正十六烷的峰,但是却出现了众多杂峰,即使条件更改,在最后一张图上,溶剂峰附近仍有较多峰挤在一起,这是什么原因造成的?是标准物质不纯?还是仪器有污染(进样前跑过一针空针,基线平整)?或是因为条件不同,出来的成分不一样,自然就有很多峰了?谁能帮解释下?
色谱很多标准品都没有验收标准,特别是中药提取出来的对照品,更是没有标准可依面对这一块的标准空缺,是不是存在严重的质量漏洞呢而更有的标准品,纯度并不是很高,你又是如何看待这个问题呢?很多标准品的质量检测报告都是归一化法,你觉得合理吗
质监局公布了300多项食品添加剂标准目录,怎么看到了很多都是陈旧的标准,而且还是N年前的标准~您说这还让人放心乎?这些食品添加剂,您做了哪些?
请问老师有证标准物质查了很多网站都没有的话,可以用RM标准物质来做溯源性吗
不太了解团体标准,但是以后可能很多行业都要制定团体标准,大家对这个怎么理解?对仪器行业有木有影响?
在行业标准区不能上传标准,求助了很多人,都没有回信,版主也不在,请求帮助,到底是怎么回事?试了好几天都不行啊
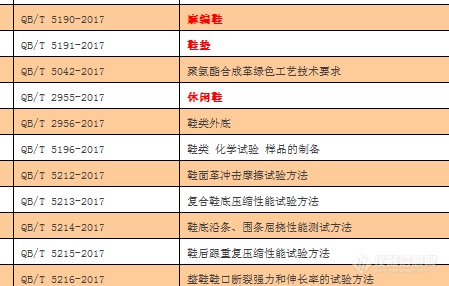
2017年出了很多鞋类标准,大家都收到标准了吗?老师分享一下吧?[img=,449,286]http://ng1.17img.cn/bbsfiles/images/2018/03/201803081059281157_1723_2154459_3.png!w449x286.jpg[/img]
购买的猪肝标准物质,锰的认定值为10.01±0.4微克/克。称取0.25克,用4:1的硝酸高氯酸混合酸消解完全后用纯水定容至25ml。(锰的理论浓度应为0.1微克/毫升)但是,反复测了很多次,都只有0.06微克/毫升左右,比理论值小了差不多一半。仪器型号耶拿NOVAA400。标准曲线浓度梯度使用6个点,分别为0.05、0.1、0.15、0.2、0.25、0.3微克/毫升,且做出来标线良好,相关系数0.9993。测定相关条件:波长279.5。狭缝0.2。乙炔流量60NL/h 。灯电流7mA。燃烧头高度6mm。雾化器提升量5ml/min。搞了几个小时,测出来的值始终达不到标准值并且相差非常大。现请教各位老师,究竟是什么原因?在此先谢过了
很多国家标准火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法上机样品溶液的溶剂用的盐酸或硝酸浓度达到5%(V/V),看很多版友和经过自己的验证发现,盐酸1%或硝酸0.5%与浓度5%比没有差异,为什么标准使用使用这么高的浓度,既浪费还污染环境,增加职业病接触风险?
原子吸收标准曲线有很多种拟合方式,我多数是用非线性过零点(使用的标液比推荐的浓度稍大点)拟合的是曲线,而不是直线。公司有人认为应该使用线性计算截距来拟合曲线,认为截距可以影响检出限。我认为标液浓度较大时积分的曲线不是呈正态分布对称状态的,会有一定的拖尾和肩峰,可能无法完全计算面积,会呈梯度降低,所以出现了非线性,系统误差同时存在,不会干扰结果(标准物质也能对上)。最小二乘法看了半天没有明白(抱歉 数学没学好)不知道这几种拟合方式有什么区别?用的是PE的仪器子。
怎么没有极谱法?催化极谱法现在还有人使用吗?很多标准方法还是用的这个。。。。。。。
是不是要备一套正版标准的呢?我们现在很多都是网上下来用的,这样行不行的呀??
请问: 用波长色散型XRF测标准样品,计数率低很多,不知道是什么问题啊?
各位哥哥姐姐们好,不知道为什么,我的标准品过针孔滤膜时峰面积会减少的很多,大约一半还要多一点,一开始我以为是我回收率的问题,可是做空白添加药物峰面积也比不过膜的标准品的峰面积要小,再后来一试原来标准品直接过膜面积就见效了很多,我考虑过膜吸附,用甲醇洗膜后基本没有效果,也考虑了用了0.45um的膜,也是不行,我的复溶液时20%的乙腈/水。望有经验的高手能给小弟指条明路,不胜感激。
对羟基苯甲酸甲乙丙酯的标准溶液有很多杂峰正常吗?我感觉理论上应该是只有三个大的色谱峰,结果出了十多个[img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310191022364344_1417_5979722_3.png[/img]
我在做液相色谱,做标准线过程中,同个一个浓度的标准溶液进样三次,为什么波的高度不一样,面积也差很多,这是为啥?而且,峰型也有些奇怪,不是对称的,而是左侧鼓起来了,像怀孕一样,这是为啥?流速太慢的缘故吗?
现在标准品有的很多,价格很贵,如果价格能再低一些,量再大一些就好了
在用液相测组胺的时候,标准品中出现很多杂峰,请问会是什么影响因素造成的
看了很多帖子,现在已经对光谱的漂移校正和标准化模糊了,有什么联系?求解答,光谱小白一枚
用waters的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]跑标准品,但是TIC离子流图有在靠近标准品峰的位置有很多小杂峰之前我清洗过离子源了,但是好像还有杂质的样子,谱图这样是不是因为离子源或者检测器不干净呀?[img]https://ng1.17img.cn/bbsfiles/images/2021/04/202104191814554655_6160_5082352_3.png[/img]
1.检测仪器或元素:汞2.检测背景或条件:标准曲线的荧光强度值突然降低很多3.疑惑或问题:什么原因造成的呢?
这几天用岛津的碳18分析氨基酸,用的异硫氰酸苯酯为衍生剂。 用水代替氨基酸标准品,会出现很多杂峰,所以想应该是衍生过程引进的。衍生的方法是 标准品+三乙胺乙腈溶液+异硫氰酸苯酯乙腈溶液 反应一个小时候用正己烷萃取各位大虾有更好的衍生方法么?本实验室没有氮吹仪,没法吹干哦
[求助]马弗炉高温消化石墨炉法测茶叶中的铅,含量比标准值差很多!称1g茶叶,马弗炉中500度消化6h,2ml0.5mol/L硝酸溶解,然后洗入25ml比色管定容。上石墨炉。4.4(+-0.3)ppm的茶叶质控样,测量后计算得到的结果只有0.38ppm。难道是马弗炉500度太高,把铅都挥发了?
发现最近两年铝合金标准样涨价涨的厉害啊,单个就涨了二百多块钱
http://ng1.17img.cn/bbsfiles/images/2015/03/201503121043_538070_2902001_3.png请大家注意下 新标准出现了
纺织品调湿和试验用标准大气,这个在很多国标中都有要求,这个大气不是分级的吗,但是一些标准提到样品要在标准大气中调湿,没有说明几级大气,怎么办?

http://ng1.17img.cn/bbsfiles/images/2015/03/201503121043_538070_2902001_3.png请大家注意下 新标准出现了
我一直都在找红外的很多样品的标准谱图就是在网上找不到,不知道那位高手能知道在哪个个网站可以找到。谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP