标准加入法,又名标准增量法或直线外推法,是一种被广泛使用的检验仪器准确度的测试方法。这种方法尤其适用于检验样品中是否存在干扰物质。标准加入法,又名标准增量法或直线外推法,是一种被广泛使用的检验仪器准确度的测试方法。这种方法尤其适用于检验样品中是否存在干扰物质。当很难配置与样品溶液相似的标准溶液,或样品基体成分很高,而且变化不定或样品中含有固体物质而对吸收的影响难以保持一定时,采用标准加入法是非常有效的。将一定量已知浓度的标准溶液加入待测样品中,测定加入前后样品的浓度。加入标准溶液后的浓度将比加入前的高,其增加的量应等于加入的标准溶液中所含的待测物质的量。如果样品中存在干扰物质,则浓度的增加值将小于或大于理论值。标准曲线法适用于标准曲线的基体和样品的基体大致相同的情况,优点是速度快,缺点是当样品基体复杂时不正确。标准加入法可以有效克服上面所说的缺点,因为他是把样品和标准混在一起同时测定的(“标准加入法”的叫法就是从这里来的),但他也有缺点就是速度很慢。标准曲线法可在样品很多的时候使用,先做出曲线,然后从曲线上找点,那样方便。标准加入法,适合数量少的时候用。其具体操作方法是:取4—5份相同体积的被测元素试液,从第二份起再分别加入同一浓度不同体积的被测元素的标准溶液,用溶剂稀释至相同体积,于相同实验条件下依次测量各个试液的吸光度,绘制出标准加入法曲线。将此曲线向左外延至与横坐标交点Cx即为待测元素的浓度。将试液的标准加入法曲线斜率和待测元素标准工作曲线斜率比较,可说明基体效应是否存在。
原子吸收光谱法是一种相对测量法,必须采用校准的方法来获得未知样品中待测元素的浓度。校准方法是否准确,取决于待测元素在分析样品和校准溶液中是否具有完全相同的分析行为。一旦由于样品中的共存物影响了待测元素的分析行为,使之不同与校准溶液中该元素的行为,则可能使完全相同浓度的溶液给出不同的吸收值,引起干扰。 如果对干扰不够重视,未采取相应的消除措施,往往使测定结果不准确。在原子吸收光谱分析中,常采用标准加入法来抵消干扰,减少分析误差。然而,如果对标准加入法应用不慎,将会引起严重的分析误差,本文将对该法的局限性作一探讨。 校准加入法是将不同量的标准溶液分别加入数份等体积的试样溶液之中,其中一份试样溶液不加标准,均稀释至相同体积后测定(并制备一个样品空白)。以测定溶液中外加标准物质的浓度为横坐标,以吸光度为纵坐标对应作图,然后将直线延长使之与浓度轴相交,交点对应的浓度值即为试样溶液中待测元素的浓度。标准加入法的曲线如图1所示。图x的绝对值即为测定溶液中被测元素的浓度。 在原子吸收分析时,用标准加入法一般须满足三个条件:第一,待测元素浓度从零至最大加入标准浓度范围,必须与吸光度值具有线性关系,并且标准曲线通过坐标原点。第二,在测定溶液中的干扰物质浓度必须恒定。第三,加入标准物质产生的响应值与原样品中待测元素产生的响应值相同。
最近打算新扩制盐工业钙和镁的标准,标准要求用标准加入法,不知道采用标准加入法的标准该如何进行验证(线性,精密度,加标回收,检出限)等,一条曲线只能出一个值吧?难道要重复做多条曲线吗?
有色谱定量分析中,有标准加入法(standard addition method),标准的加入量是如何确定的呢?
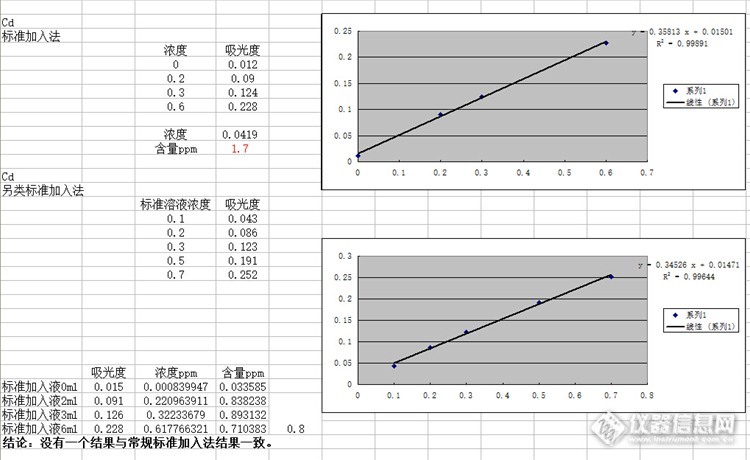
常遇到待测样品的吸光度在标准曲线的下限左右,如铅的下限0.5-1ppm,而线性的中部一般在5ppm左右,在不考虑基体干扰的情况下,此种情况在待测样品中加入4ppm的标准品,结果=(测得总量-标准加入量)/样品称样量,这样会不会比直接测定更准确?经过验收,这个方法无法克服基体干扰。标准加入法:称取10.0038g氧化锌样品→200ml容量瓶,吸取4份25mL分别置50mL容量瓶中,再分别加入0.00mL、2.00mL、3.00mL、6.00mL标准中间液(Pb:50ug/ml,Cd:5ug/ml)另类标准加入法:将上述标准加入法试样液作为样品液;分别吸取0 mL,1 .0 mL,2 .0 mL,3.0 mL,5.0 mL,7 .0mL标准中间液,置于50mL容量瓶作为标准工作液http://ng1.17img.cn/bbsfiles/images/2015/09/201509011724_564002_1638724_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509011724_564003_1638724_3.jpg
跪问各位利用标准加入法消除基体效应的具体操作步骤?将待测样品平均分成几份,然后按一定浓度梯度向里面加入标准样品,在分析时是不是就不用标准溶液了,直接测量,在分析的过程中怎么填写实际重量,实际体积,稀释系数呢?
用标准加入法是直接得结果,问用标准加入法 标准化后我直接测未知样是不是和标准曲线法一样的?没怎么做过。
请教用标准加入法检测的利弊?因为我们的样品基体抑制严重,所以用标准加入法,有谁有这方面的经验呢?[em61] [em61] [em61]
请问标准加入校正法和标准加入法的区别,标准加入法可以自动算出样品的结果,那标准加入校正法是怎样得出结果呢,哪位大侠能否告诉一下,标准加入校正法的具体操作方法,谢谢了
关于标准加入法还有几个问题请大家一起讨论一下。1.标准加入时相隔的标液浓度是如何确定?是不是与溶液中待测金属的浓度差不多。2.标准加入法都过空白,是不是不作空白也可以?是不是空白不参与计算?
在色谱定量分析中,有标准加入法(standard addition method),标准的加入量是如何确定的呢?
标准加入法是不是就是内标法?EN71-3可以用标准加入法不?
标准加入法是不是就是内标法?EN71-3可以用标准加入法不?
一种未知浓度的样品,通过极谱法测定其浓度,可以采用标准加入法吗?但是这种样品我们又没有标准物质,
请问各位老师,做硬脂酸镁镉盐限量检查时,用标准加入法。是不是这样的:取四份含同样多供试品溶液,其中一份不加标准液,另三份加不同浓度的标准液(标准液不含供试品)测?但是为什么药典里配制标准液里有供试品?
标准加入法的作用及优点?
标准加入法
为什么标准加入法测定时,还要先用已知的标准形成线性,然后再测,我认为已经试样中加入了标准,直接测为什么不可以,请大家帮助
请教各位老师,原子吸收标准加入法方法学验证中,没有已知元素含量的标准样品,准确度该怎么做?(PS:1.方法是标准加入法。2.只有样品,没有已知元素含量的标准样品。)
由于在测试铁基体样品时铅220很小(几十PPM),而铅283很大(几千PPM),我用标准加入法消除干扰,但发现标准加入法的结果中铅220仍然远小于铅283(10E-6PPM:10E-3PPM),还相差1000倍,不知为何?其中相关系数220约为1.00,283有3个9吧。另外,按标准加入法的一般做法,加入的标液浓度应与样品浓度差不多(在一个数量级上吧,我听仪器商说的),但我若取该样品铅含量为ND(10E-6PPM),而我加入的标液浓度是2PPM,5PPM,10PPM,样品浓度与加入的标液浓度相差太大,我怎么感相信结果呢?
引进一台新极谱884,厂家设置计算方法为标准加入法,过程是,先加入空白溶液,扫描,然后加入待测样品,扫描得峰高,这样合理吗
标准加入法由于可以抑制基体的影响, 抵消干扰, 减小分析误差等特点, 现已广泛应用于[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析中。在难于制备可以代表样品的标准溶液时, 这个方法尤为适用。但是,标准加入法也存在很多的问题。 1、背景吸收问题如果测定中存在背景吸收, 将使横轴向上移动。必须先进行背景校正, 否则, 标准加入法将出现错误结果。2、基体干扰问题如果干扰程度随测定元素与干扰元素的比值而变化, 标准加入法将不能被采用。例如, 在有磷酸盐和铝存在时测钙含量, 由于形成了稳定的络合物或不完全挥发物, 出现干扰程度随测定元素与干扰元素比值变化而改变的现象。3、元素存在形式问题在石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析中, 同一元素形成的不同化合物或不同价态之间很可能有不同的分析行为。最常用的基体改进剂硝酸镍, 对不同价态的硒稳定程度不同, 如果作为标准加入的硒为四价的亚硒酸盐, 而样品中的硒为二价的硒化物,这种情况下如果采用硝酸镍作基体改进剂, 所得结果在很大程度上取决于加热程序。由此可见, 遇到这种情况, 标准加入法无济于事。在石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析中, 要克服使用标准加入法的局限性, 需要采用更有效的元素形态和加入合适的基体改进剂。4、 电离干扰问题由于电离干扰导致分析曲线弯向吸收轴, 该干扰不能靠标准加入法校正, 唯一的校正方法是加入阻电离剂, 如氯化铯等。总之, 在某些合适的情况下, 标准加入法在[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析中是卓有成效的, 但也需要谨慎, 否则会引起严重的分析误差。
弱弱的问一下,对于标准加入法这个概念很不清楚,书上说:“将校正曲线外延与横坐标相交,原点至交点的距离即为被测元素含量”,这句话怎么理解? 还有pe800如何编辑标准加入法,
加和性干扰如果测得的吸光度A中,除了待测元素的吸收B外,还有一个附加的吸收C,且C不随待测元素的浓度而变化(C值可以是正的,也可以是负的),则这种干扰为加和性干扰。加和性干扰的特点是不改变曲线的斜率和形状,只改变曲线的在吸收轴上的截距。光谱线干扰、背景吸收和污染等一般可归于加和性干扰。加和性干扰采用标准加入法是消除不了的。测得的吸光度A=B+C,无论如何加入已知浓度的待测元素,C值是始终消除不了的。校正光谱线干扰和背景吸收的方法是配制尽可能与样品相同基体的标准系列,同时进行背景校正。校正污染需使用完全与样品一样,经过相同前处理的空白作参比。特效性干扰在标准加入法中,加入元素和待测元素从表面上看是同一元素,两者又同处于相同的环境,似乎应该具有完全相同的分析行为。但是,问题并非如此简单,在一定的条件下,即使相同的物种、相同的化合物也可能有不同的分析行为,常常表现为不同浓度的分析元素受到的干扰程度不同,这称为特效性干扰。在火焰原子吸收中,全部的电离干扰和部分的化学干扰都属于特效性干扰,不能通过标准加入法来消除。电离干扰可加入消电离剂(电离电位低元素)等方法来抑制,而特效性的化学干扰可加入稀放剂、保护剂等方法来抑制。在石墨炉原子吸收中,许多基体干扰(多数的蒸发干扰和全部气相干扰)是特效性的,不能通过标准加入法来校正。例如,如果标准溶液中的待测元素是以不挥发的无机盐类的形式存在,而样品中的待测元素是以较挥发的有机化合物的形式存在,那么很可能在挥发阶段样品中的待测元素挥发损失了一部分,而标准中的待测元素留下了,显然结果不可能正确。这部分的干扰应通过异构重整或基体改进技术加以抑制。
各位在用标准加入法的时候是做几个点,大概要加的浓度是怎么定的,依次是多少
大家好,今天我配了两个标液一个是10ppb的铅 一个是50ppb的铅,做了一下标准曲线法和标准加入法的对比。 试验一:我用50ppb的铅 做了一条标准曲线 15 ,20,30,40,50做了一标准曲线,相关系数0.999 测量10ppb的铅,得到的结果是11.2ppb 试验二:我用50ppb的铅当成储备液,把10ppb的铅当成未知样品,进行标准加入法测量,做了三个点,第一个点加入10ppb,第二个点加入20ppb,第 三个点加入了30ppb,相关系数是0.9989,得到的blank(就是10ppb的样品)结果是9.1ppb 试验三:我用50ppb的铅当成储备液,把10ppb的铅当成未知样品,进行标准加入法测量,做了五个点,第一个点加入10ppb,第二个点加入20ppb,第三个点加入30ppb,第四个点加入40ppb,第五个点加入50ppb,相关系数是0.999,得到的blank(就是10ppb的样品)结果是10.1ppb备注:试验一样品取20微升,稀释液10微升,基体改性剂5微升 试验二和试验三样品取10微升,稀释液取10微升,基体改性剂取5微升。问题一:标准加入法和标准曲线法测同一个样出现了三个不同的结果(9.1,10.1,11.2),是什么原因造成的? 标准加入法应该至少做多少个点才比较合适?是不是点越多越好? 标准加入法的相关系数是不是要一般要达到多少才可以出数据?不确定度如何评估? 这几个数据是不是都是正常的?对于试验一的数据,我怀疑是不是标曲的问题,第一个点15是不是有点偏高?不过要标准自动取样器不少与五微升,所以第一个点也只能取15ppb,由于不是落在中心,所以结果的偏差比较大?
请问大家,常量组分的定量分析中,能否使用标准加入法进行定量(例如某组分的含量在20%-70%间,且已经验证在1%-99%均有良好的线性关系,这种情况下,可以用吗?可能大家觉得这种分析外标法就好,但就标准加入法而言,是否可行)?如果适用的话,一般标准物加入的量大概是多少,有什么要求,有老师做过类似的分析没?另外,标准加入法公式中w=Δw×A/ΔA(w是组分量,A是峰面积,Δ是变化量),资料上说,标准加入法对进样量要求不必十分准确,进样量稍有区别影响不大,但是根据公式要计算ΔA,若两次进样量不严格一致,那么峰面积的改变量就不是一个确定值(因为进样量直接决定峰面积,进样量大,峰面积就大),所以我认为,两次进样量应该完全一致,大家怎么看?再有,标准曲线法中,A=a+bw,通常我们会根据相关系数的大小来确认线性关系,一般是0.999或以上即可,不知道平常大家有没有关注这个a值的大小,在定量分析中需要验证下a值么(就是a值在什么范围内才是合适的,此时仪器的系统误差在合理范围内;资料上就说a值越接近0越好,具体多少没写),如何验证的?
标准加入法的操作中,要求待测溶液的体积相同,再按比例加入一定浓度的待测物标准溶液,进行定容。现在有一个问题,假如我使用原液进行定容,那么就不能保证待测溶液的体积相同,如果是同高纯水定容,那么他们的稀释倍数怎么计算?
看了《色谱定性与定量》一书,中间讲到标准加入法的优缺点时,有这么一段话“标准加入法的优点是.....进样量不必十分准确.....”。我就不明白了,如果标准加入前和加入后的进样量不一样的话,那有可能加入了标准那次出的峰面积还没有不加标准出的峰面积大,那样怎么进行下一步的计算?
在我的机器的说明书上看到,消除基体干扰的最好方法是标准加入法,但基体干扰至少包括背景干扰和光谱干扰(光谱干扰又包括部分谱线重叠,完全重叠)啊,标准加入法能消除这两种干扰吗?我做了一个标准加入法的实验(在铅标准液中混入大量铁,测试标准加入法能否消除铁对铅283的干扰),数据并没有显示该方法消除了光谱干扰.大侠指教







 我要推广仪器
我要推广仪器
 下载APP
下载APP