求助草甘膦检测条件。按照国标很不稳定
我用安捷伦的1290-6460检测茶叶中的草甘膦、氨甲基膦酸,标准是SN/T 1923,可是母离子、子离子都找不到啊,麻烦大家指点一下!
草甘膦检测的优选法是用什么仪器做?水中草甘膦我用液相实在是做不出来了,有哪些方面应该注意,望老师不吝赐教
[color=#444444]80%草甘膦.草铵膦可溶粉(20:60)如何用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、液相色谱或者其他方法检测?急需具体详细的检测方法。[/color]
茶叶中草甘膦的检测,加水是用来提取,加二氯甲烷的作用是什么?
我们一直用气质检测茶叶中草甘膦。但由于要做衍化,前处理比较复杂。为此还专门购进能制冷零下60度的超低温冰箱。文献上草甘膦的检测大部分要用衍生化。随着LC-MS/MS的推广,已经有专家研究在LC-MS/MS上不用衍生化检测草甘膦的方法。我们今年也购进了AB的Triple Quad 4500。我们想利用手中的这一利器,研究不用衍生化检测茶叶中草甘膦的方法。不知哪位同行有相关的资料?
有做草甘膦检测的老师吗,想咨询一下我们前面草甘膦只出一个峰响应也好,后面突然变成出两个峰了,而且其中一个子离子的响应变很差,是什么原因引起的?怎么解决呢?[img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906180934400959_7320_3490692_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906180934399592_8297_3490692_3.png[/img]
小白求问,在使用液相质谱进行草甘膦项目的检测中。衍生化实验使用FMOC CL说是对氨基进行保护,那么保护氨基的目的是什么呢?是提高分子量排除小分子干扰还是有其他原因?

农药残留草甘膦的检测前言 草甘膦又叫镇草宁,是广谱除草剂,对哺乳动物有低毒性。除草剂的广泛使用使得草甘膦成为地下水甚至饮用水的重要污染物。简介 分析草甘膦的方法现在主要有气相色谱法或液相色谱法,通常都要衍生化处理,其中采用液相色谱法更多些。现主要介绍比较复杂的阳离子交换柱后衍生荧光检测器测定草甘膦的方法。本方法着重描述了分析草甘膦及其主要代谢物氨甲基磷酸(AMPA)的便利方法。 本方法参考了美国EPA方法、GBT 5750.9-2006方法、岛津、美瑞泰克、博纳艾杰尔等公司提供的参考方法及本实验室全体工作人员的集体智慧编制而成。阳离子交换柱后衍生荧光检测器法测定草甘膦色谱原理 柱后衍生与荧光检测器结合使得本方法具有很高的灵敏度和选择性。样品进样后,草甘膦和AMPA经流动相磷酸二氢钾缓冲液洗脱,在色谱柱中分离。分离后,分析物通过柱后衍生反应系统,生成的反应产物用荧光检测。由于每个厂家的仪器的灵敏度各不相同,所以每种方法的进样量也不相同,一般为10-200ul.此方法建议定量进样50uL,如图1所示,效果很好。http://ng1.17img.cn/bbsfiles/images/2013/09/201309200103_465474_2369266_3.png图11.草甘膦 1.0mg/L 2.AMPA 1.0mg/L设备 L600高效液相色谱仪,包括:梯度泵1套,柱后衍生仪1台,衍生注射泵2台,荧光检测器1台,数据采集控制器1台,自动进样器1台,柱温箱1台,阳离子色谱柱及保护柱各1根,色谱工作站1套,超声波振动仪,溶剂过滤器,固相萃取装置,离心机。试剂 5 mM 磷酸二氢钾,磷酸调pH 2.0 ;5 mM 氢氧化钾 ;5%次氯酸钠溶液;硼酸钠缓冲稀释剂;OPA(二巯基乙醇),色谱级;甲醇,色谱级;硫醇试剂。试剂和标准品的准备氧化试剂(试剂1): 将一瓶次氯酸盐稀释剂倒入预先用甲醇清洗过的干净试剂瓶中。加入100uL5% 次氯酸盐溶液(家用漂白剂),旋转,混匀,0.45um水系滤膜过滤,超声脱气。加入的剂量可能需要根据检测器的响应进行调整:色谱系统平衡后,草甘膦混合测试溶液进样10uL,如图2所示。如果AMPA和草甘膦的面积比例差别很大,则在氧化剂里面再加入5%次氯酸盐溶液,每次加入20uL,直至二者所占面积比例相当。 OPA试剂(试剂2): 将OPA稀释剂倒入预先用甲醇清洗过的干净试剂瓶中。鼓泡10min以除去其中的氧气。余下的步骤应在尽可能短的时间内完成,因为此试剂对氧气和光敏感:称取大约100 mg OPA于一小烧杯中,加入10 mL甲醇溶解,而后加入OPA稀释剂。用1~2 mL甲醇清洗烧杯,把清洗液倒入稀释剂中。加入2 g硫醇试剂于试剂瓶中。0.45um有机系滤膜过滤,超声脱气,备用。色谱条件分析柱:草甘膦分析柱,阳离子交换柱,4 mm x 150 mm x 8um保护柱:草甘膦保护柱,阳离子交换柱,3 mm x 20 mm x 8um柱温:55 °C 淋洗液:(A)磷酸二氢钾(5 mM,磷酸调pH 2.0);(B)氢氧化钾(5 mM)流速:0.4 mL/min 柱后试剂1:氧化试剂,36 °C,流速为0.4 mL/min 柱后试剂2:OPA,室温,流速为0.4mL/min 荧光激发波长为330 nm,发射波长为460 nm 进样量:50ul剃度表:[fo
按照各位大侠的意见,我查了一些资料,好像草甘膦、百草枯等农药极性很大,而且是经常检测的农药。我想问问这些农药残留用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]检测的可行性有多大?哪位大侠有相关的资料能否分享一下!谢谢!
求助,食品中草甘膦的检测方法,我们参照SN-T 1923-2007 进出口食品中草甘膦残留量的检测方法 液相色谱-质谱 质谱法,做出的回收率很低,请问大家都是用什么方法来检测的?有没有用气相来做的?查到了2个气相的标准。请大家给点帮助,谢谢!http://simg.instrument.com.cn/bbs/images/brow/em09511.gif
各位高手:我现在遇到很棘手的问题,希望大家帮帮忙,着急啊。草甘膦含量95%以上,杂质含量0.5%以上的都要能够检测出。杂质包括:甘氨酸,亚氨基二乙酸,双甘膦,增甘膦。整个检测时间不超过15分钟,并且检测方法不能太复杂。检测器可为紫外或者DAD或ELSD或其他都可以。希望各位高手指点指点阿。我都快急疯了。谢谢阿。
茶叶中农残检测中草甘膦的检测方法。。。感兴趣的可以联系我。
谁有草甘膦残留检测方法?
自来水7月1日实行新国标,特把自来水中草甘膦、胺甲基硫酸的检测方法拿来共同分享
最近要检测草甘膦,不知道有没有做过的,我们现在没有离子交换柱,若干试剂,有没有做过的朋友交流下。
哪位大神提供一下银杏叶中草甘膦残留的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]或液相色谱检测方法。
水中草甘膦用[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]检测有哪些方法?哪个方法前处理简单不容易出问题?
分享资料《柱前衍生-超高液相色谱-串联质谱法同时检测茶叶中草甘膦和草铵膦的残留量》http://wenku.baidu.com/link?url=meaZziIax7otjPVs1FrYqEHoNBY-97XuQ_wZBaHHvfC7ZpBS0qhoL2Oqu-x9ExmGEBRNVW6stKsrlDk7gULF6T_eXev3FAk3Uwgt6BdAVT7
看到一篇《[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]法检测土壤中草甘磷、草铵磷及其代谢物》的文献,分享给大家。近一段时间看到网友提出草甘磷的检测出现问题,正好有这样一篇,看看是否有启示。
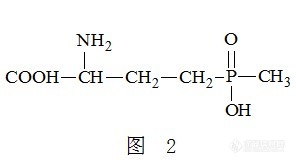
关于”新版GB 2763 食品安全国家标准 规定茶叶中限量农残草甘膦和草铵膦项目“的检测研究 一、研究意义及现状 随着新版GB 2763 食品安全国家标准的不断更新及发布实施,草甘膦和草铵膦已被明确列为茶叶中农药残留强检(必检)项目,草甘膦在茶叶中的限量为1mg/kg,草铵膦在茶叶中的限量为0.5mg/kg。同时,草甘膦和草铵膦也成为中国茶叶出口国外的检测项目(来源于中华人民共和国商务部),且已成为越来越严的限量指标。 文献(2013年农药行业预测和草甘膦市场机遇分析,杨益军,农药市场信息,2013.03)报道,除草剂草甘膦因其高效、广谱、低毒等特性使其被广泛应用,未来需求量也将大幅增加。但草甘膦的使用容易使植物产生抗性(IARC国际研究机构发布报告称草甘膦很可能对人类致癌),而草铵膦可克服该缺陷,现已有学者(草铵膦、百草枯、草甘膦对非耕地杂草的防效比较,凌进,农药,2014年第53卷第8期,613-615)对草铵膦和草甘膦的除草性能进行了研究,确证了草铵膦代替草甘膦的可行性。 因草甘膦和草铵膦为广谱除草剂,被广泛应用于农业、林业及园艺的栽培。我国作为农业大国,其茶叶产量世界第一、出口量世界第二,草甘膦和草铵膦的生产和使用量都位居世界前列(草甘膦 草铵膦及其代谢产物的检测方法,李小娟、周信康、孟品佳,公共安全中的化学问题研究进展)。同时,我单位对西南茶叶原料主产区进行了初步调研,进一步确认茶农使用草甘膦和草铵膦农药的现状。 随着草甘膦和草铵膦除草剂使用量的日益增大,使其常被发现存在于环境水样、土壤及植物中,这样长期积累会引起环境污染,从而对人类健康造成严重威胁。草甘膦和草铵膦结构类似,且均含有膦酸基、羟基、氨基,是极强的两性化合物,易溶于水,难挥发。鉴于草甘膦和草铵膦特殊的物化性质和茶叶基质自身的复杂性,无论国内外,茶叶中草甘膦和草铵膦同时检测的标准还未见发布。 目前,可用于检测草甘膦和草铵膦农药残留量的主要方法有液相色谱法,柱前衍生后气相色谱法、气相色谱-质谱法及液相色谱-质谱/质谱法。 快速发展起来的超高效液相色谱-质谱联用技术,具有检测灵敏度高、适用范围广、分析速度快和能有效排除复杂基质产生的干扰等优点,当今已成为检测型实验室检测农残的首选。然而,若采用液质质直接测定草甘膦和草铵膦,则仪器响应较低,无法满足茶叶中草甘膦和草铵膦农药残留量检测的要求。 近两年来,已有研究文献陆续发表,用柱前衍生-液相色谱串联质谱法检测。笔者结合其文献研究结果,对茶叶中草甘膦和草铵膦农药残留量的检测方法系统地进行研究,采用9-芴甲氧羰酰氯(FMOC-Cl)作为常用衍生剂,在硼酸盐缓冲盐溶液的条件下,能与草甘膦和草铵膦的提取液发生衍生反应,形成衍生产物,衍生产物注入UPLC进行色谱洗脱分离,采用串联质谱探测响应信号,外标法直接快速定量茶叶中的草甘膦和草铵膦的含量。二、液质质检测分析原理 质谱原理是先将物质离子化,按离子的质荷比分离,然后测量各种离子谱峰的强度而实现分析目的的一种分析方法。液质联用是将色谱的分离能力与质谱强大的定性功能结合起来,实现对复杂混合物更准确的定量和定性分析,简化样品的前处理流程,使样品分析更简便。主要针对不挥发性、极性、热不稳定、大分子量等化合物的分析测定。液质联用检测技术灵敏度高,且串联质谱(三重四级杆)定性准确,可有效杜绝微量甚至痕量物质分析时的假阳性现象,常用于目标物质的痕量分析。 采用柱前衍生-液相色谱串联质谱法检测茶叶中的草甘膦和草铵膦有以下优势: 1)、灵敏度高、线性好、检出限低(可达ng/mL级及其以下); 2)、定量结果准确、稳定、重复性好; 3)、实验操作简单、步骤少、耗时短、分析速度快、检测效率高; 4)、实验试剂无污染、无毒、安全; 5)、有效减弱基质对目标物检测的影响。三、茶叶中草甘膦和草铵膦农药残留量检测的前处理试验 茶叶经GB/T8303磨碎、过筛制得待测茶样(发酵茶应先低温去除水分,使样品易于磨碎); 准确称取已磨碎处理过的茶样1g(精确至0.001g)置于80mL具盖离心管中,加入10mL水,涡旋混匀静置,加入2mL二氯甲烷,混匀,超声提取或回旋振荡10min,低速离心机4500r/min离心5min,取上清液,制得提取上清液; 注意:若茶样为新采摘的鲜叶,则称取约5g鲜叶于研钵中,加入30mL水,研磨约10min,将其转入离心管中,用10mL水洗涤研钵后转移至离心管,重复洗涤一次,再次加入10mL二氯甲烷于离心管,均质至混匀,4500r/min离心10min,取上清液,制得提取上清液; 将提取上清液用净化柱CAX、C18,以及活性炭小柱等进行比对试验,确定以C18小柱净化提取液,制得提取净化液; 通过缓冲液浓度、衍生液浓度、衍生液用量、缓冲液用量、净化液用量,衍生时间等条件试验,得出最优衍生试验参数为缓冲液浓度为50g/L,衍生液浓度20g/L,衍生液用量:缓冲液用量:净化液用量的体积比为1:1:1,衍生时间为约3h,衍生液过0.22μm的有机滤膜后进样。四、茶叶中草甘膦和草铵膦农药残留量检测的衍生机理 茶叶中草甘膦和草铵膦农药残留量的衍生机理为:在硼酸钠缓冲盐溶液条件下,草甘膦(分子结构如图1所示)和草铵膦(分子结构如图2所示)中R-NH-R’的-H被FMOC-Cl(分子结构如图3所示)中的FMOC-取代,生成 http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414494663_01_0_3.png,得到衍生目标产物草甘膦衍生物和草铵膦衍生物。 其中,草甘膦分子结构图,见图1;草铵膦分子结构图,见图2;9-芴甲氧羰酰氯(FMOC-Cl)分子结构图,见图3;草甘膦和草铵膦与9-芴甲氧羰酰氯(FMOC-Cl)的衍生机理图,见图4。http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414424276_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414430242_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414432007_01_2275853_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507061123_553629_2275853_3.png五、茶叶中草甘膦、草铵膦农残衍生物在质谱中的裂解机理1、茶叶中草甘膦农残衍生物在质谱中的裂解机理 通过对草甘膦衍生物在串联质谱中的裂解机理进行系统的分析研究,可探索出草甘膦衍生物的裂解机理为:首先草甘膦衍生物裂解为离
[color=#444444]我最近正在做草甘膦的检测,柱前衍生了,结果质谱摸方法找不到对应的离子,求助,大家看看有什么错误吗?[/color][color=#444444]取1mL样品溶液, 分别依次加入 1. 0mL 50g/L硼酸钠缓冲溶液、 1. 0mL 20g/LFMOC-Cl 丙酮衍生液, 混匀后室温下衍生 2h, 以 4500r/min 转速离心 5 min, 过 0. 2 μm 有机微孔滤膜后,供超高效液相色谱- 串联质谱测定。[/color]
最近要做食品中草甘膦的检测,在站内查到了农业部的一个标准,NY/Y 1096-2006,前处理是用水提取的,我准备直接用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测,省去了衍生这一步。不知道还有没有其他前处理的方法?
各位老师,我一做农药检测的客户,现在要做草甘膦检测,用的是GBT 23750-2009 里面用的CAX小柱不知道在哪里找,求助各位大神,有没做过该项目的,帮忙找找。谢谢!
用GC-MS检测草甘膦标样和样品都需要衍生化。标准样的衍生化可以用100ppm,50ul进行衍生,然后再用衍生后的来稀释至各需要浓度么?还是只能标曲中的每个浓度都进行衍生?我认为应该是可以的,但没验证过。不知哪位大虾能不吝赐教?谢谢!另外,标样的浓度如何计算?比如,标准样用100ppm,50ul进行衍生,最后定容至500ul,其浓度应为10ppm,对吧?如果进行加标回收试验,如在12.5g样品中加100ug标样,加入125ml水,浸提后,取上清液20ml,加入15ml二氯甲烷萃取,离心。取水相的上清液4.5ml,加入0.5ml酸调节液混匀。取1ml 过SPE,最后定容至2ml。取浓缩液50ul进行衍生,最后定容至500ul。请问:最后定容液中标准样的理论值应是多少?是不是0.036ug/ml? 谢谢!

最近对茶叶、大豆粕中草甘膦的检测进行了优化,采用一种新的净化方式,净化液直接加入衍生试剂FMOC-CL,[color=#cc0000]Primer[/color][color=#333333] [/color][color=#cc0000]XE [/color][url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS分析,定量限达到10ug.kg。有需要交流的朋友可以QQ402179727。茶叶样品净化后的照片:[img=白色为茶叶样品该方法净化后的效果,690,517]https://ng1.17img.cn/bbsfiles/images/2018/12/201812111103138250_7350_1643455_3.jpg!w690x517.jpg[/img]
液相色谱检测土壤中残留的草甘膦,我使用的紫外检测器和强离子交换色谱柱,一种是柱前衍生后检测使用C18色谱柱,一种是直接用强离子交换色谱柱做,但是都不行检测的效果不好,直接用强离子交换色谱柱做效果要好一些但是检测的极限不够,如果哪位同行做个这个指点指点啊!
想问一下,哪里可以监测土壤及水样中的草甘膦含量?
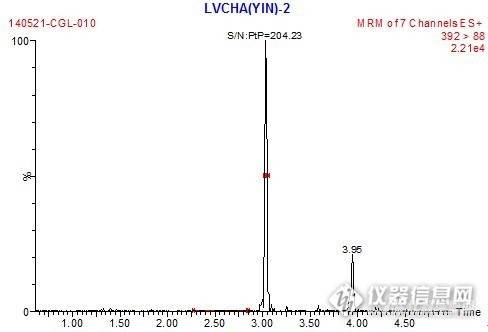
【生活中的仪器分析】样 品:蔬菜、水果、茶叶、茶粉等食品检测项目:草甘膦、草铵膦、氨甲基膦酸参考标准:SN/T 1923-2007检测仪器:a.WATERS液相色谱串联质谱仪:配有电喷雾(ESI)离子源(可用其他品牌作用等效的高效液相色谱质谱仪替代)b.Biotagevacmaster固相萃取仪c.IPRE Qclean PMG草甘膦专用固相萃取柱d.BiotageTurbovap LV 快速浓缩仪e.IKA MS3 涡旋混匀器g.TOMY-MX307离心机g.昆山超声波清洗器实验过程:1.提取及预处理称取2-5g(精确到0.001g)试样于50 mL聚丙烯离心管中,加入100μL内标液,加入20.0 mL水超声提取30min,于10000 r/min离心5min,取1.0 mL上清液于2mL子弹头离心管中,加入100μL酸度调节剂(注A),涡旋混匀,15000r/min离心5 min,待净化。注A:酸度调节剂配制方法:纯水+色谱纯甲醇+盐酸=160+40+13.4(V/V/V)2.固相萃取净化І将PMG-І柱(蓝柱)用2mL甲醇和2 mL 0.5%甲酸淋洗活化并自然滴干,将加入酸度调节剂处理的提取液(2)转移到小柱上,用5mL刻度试管收集流出液(1-2滴/秒),用1.0mL 0.5%甲酸洗柱并真空抽干,合并流出液,用移液枪吸取50%NaOH调pH7-9(用1-14pH试纸,根据样品不同约20-50μL),加水定容到3 mL刻度,混匀,待衍生。3.衍生步骤准确吸取600 μL净化液(3)于2mL子弹头离心管中,加入200 μL 5%硼砂溶液,边涡旋,边加入200 μL 25g/L FMOC-Cl乙腈溶液(注B),放置10min,加入50 μL甲酸,涡旋混匀,15000 r/min离心5min,吸取上清液准备过PMG-ІІ柱。注B:25 g/L FMOC-Cl乙腈溶液配制方法:称取0.25 gFMOC-Cl,溶解于10 mL色谱纯乙腈中。4.固相萃取净化ІІ 将PMG-ІІ柱(红柱)用2 mL甲醇和2 mL 0.5%甲酸淋洗并自然滴干,将上清液过PMG-ІІ柱,用3 mL水淋洗小柱,真空抽干5-10 min,再加入2 mL正己烷淋洗小柱,滴干后真空抽干5 min,最后用5 mL 5%氨水/甲醇洗脱小柱(1-2 mL/min)并用5 mL刻度试管收集流出液,45℃,氮气吹至近干,用20%乙腈定容1.0 mL,涡旋混匀,过0.2 μm PTFE膜后上机测试。5.测定5.1色谱条件a.色谱柱:Waters BEH-C18,1.7 μm,2.1 mm×100 mm;b.流动相:5mmol/L乙酸铵:乙腈梯度洗脱,梯度表见表1; 表1 流动相及梯度 时间(min)流速(mL/min)5mmol/L乙酸铵(%)乙腈(%)00.3901020.362384.40.362384.50.35956.50.35956.60.390109.00.39010c.检测器:串联四极杆质谱仪;d.柱温:35℃;e.进样量:10 μL。5.2质谱分析条件a)电离源:电喷雾正离子模式;b)毛细管电压:3.50KV;c)源温度:120℃;d)脱溶剂气温度:400℃;e)脱溶剂气流量:700L/h;f)碰撞室压力:2.7í10-3mbar;g)特征离子及参数见表2。 表 2 草甘膦和氨甲基膦酸的主要特征离子 化合物保留时间(min)母离子+(m/z)锥孔电压(V)子离子(m/z)碰撞能量(eV)草甘膦1.32392.215*88.02515214.01
请问 哪位老师做过草甘膦和氨甲基膦酸[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS检测,求前处理和仪器检测方法?







 我要推广仪器
我要推广仪器
 下载APP
下载APP