常常看到一个小小的维生素E标准品,竟然有好多相关标准品,如维生素E/生育酚、α-维生素E、β-维生素E、γ-维生素E和δ-维生素E、维生素E乙酸酯、维生素E棕榈酸酯、维生素E琥珀酸酯、DL-α-维生素E 标准品、DL-α-维生素E醋酸酯 标准品,太多了,我要做食品里面的维生素E的含量检测,到底要选哪个是合适的?http://simg.instrument.com.cn/bbs/images/brow/em09506.gif
那儿有a β γ δ维生素E和三烯酚的标准买啊?E
最近做食品中维生素B1的检测,依据GB5009.84-2016,高效液相,按要求配制的一系列浓度的标准液,竟然会有过载的现象,各个浓度峰面积也大概高了一倍,不知道是怎么回事,请有经验的专家帮忙解答就下3.3 标准品维生素B1 标准品:盐酸硫胺素(C12H17ClN4OSHCl)),CAS:67-03-8,纯度≥99.0%。3.4 标准溶液配制3.4.1 维生素B1 标准储备液(500μg/mL):准确称取经五氧化二磷或者氯化钙干燥24h的盐酸硫胺素标准品56.1mg(精确至0.1mg),相当于50mg硫胺素、用0.01mol/L盐酸溶液溶解并定容至100mL,摇匀。置于0℃~4℃冰箱中,保存期为3个月。3.4.2 维生素B1 标准中间液(10.0μg/mL):准确移取2.00 mL 标准储备液,用水稀释并定容至100mL,摇匀。临用前配制。3.4.3 维生素B1 标准系列工作液:吸取维生素B1 标准中间液0μL、50.0μL、100μL、200μL、400μL,800μL,1000μL,用水定容至10mL,标准系列工作液中维生素B1 的浓度分别为0μg/mL,0.0500μg/mL,0.100μg/mL,0.200μg/mL,0.400μg/mL,0.800μg/mL,1.00μg/mL。临用时配制。
我们要开展婴幼儿乳制品中维生素类的测定,维生素A的标准好象有好几种,好象有维生素A乙酸酯、维生素A酸等等,请哪位高手给点意见,具体应该买哪种?
最近准备做维生素A,GB5009.82-2016中在标准溶液配制和分析结果表述中都是用维生素A表述的,分别用“准确称取25.0mg维生素A标准品”“X——试样中维生素A的含量,维生素A单位为微克每百克(μg/100g)”表述;我们买的标品是维生素A醋酸酯;我们的检验报告单又以“维生素A(以视黄醇计)”体现。我查到1IU维生素A=0.3μgRE 1IU维生素A=0.344μg维生素A醋酸酯等换算关系,但在实验过程中究竟该如何处理这些关系了,比如要准确称取25.0mg的维生素A标准品,那我该称多少的维生素A醋酸酯;维生素A醋酸酯需不需要皂化;维生素A又称视黄醇,那检验报告单中维生素A(以视黄醇计)作何理解,视黄醇和视黄醇当量有何异同;标准品和对照品有何异同等。拜托吧里大神赐教啊,万分感谢
哪位大侠有国外检测维生素(主要是B族维生素)的标准的麻烦传个上来,小弟急用
维生素B1是维生素中发现最早的一种。由嘧啶环和噻唑环通过亚甲基结合而成的一种B族维生素。为白色结晶或结晶性粉末;有微弱的特臭,味苦,有引湿性,露置在空气中,易吸收水分。在碱性溶液中容易分解变质。酸碱度在3.5时可耐100℃高温,酸碱大于5时易失效。遇光和热效价下降。故应置于遮光,凉处保存,不宜久贮。 在酸性溶液中很稳定,在碱性溶液中不稳定,易被氧化和受热破坏。还原性物质亚硫酸盐、二氧化硫等能使维生素B1失活。(摘自百度百科) 购买回的维生素B1(盐酸硫胺)纯度标准物质(BW3600,中国计量科学研究所)给出的均匀性检验和稳定性考察的前处理条件是105℃干燥至恒重后称取一定量,加入盐酸水溶液溶解,过滤上机测定。而在GB/T 14700-2002《饲料中维生素B1的测定》标准(本法用的反相HPLC)中,标准储备液是称取一定量,用25%乙醇溶液超声溶解。在GB/T5009.84-2003《食品中硫胺素(维生素B1)的测定》标准中,称取经氯化钙干燥24h的一定量硫胺素,溶于0.01mol/L中。在中国兽药典、中国药典中,维生素B1用的是约0.1mol/L盐酸溶液溶解,而其中检验干燥失重时,用的也是105℃。 疑问:根据第一段说法,VB1并不耐热,为何要用105℃? GB/T 14700-2002中用乙醇溶液溶解,储备液能保证稳定,不选用盐酸溶液,是否更便于HPLC测试?
根据GB 5413.9-2010中附录A的方法校正维生素D的浓度,用乙醇来溶解,但是没有颜色显示,我用1000ug/ml和10ug/ml的标准储备液测出来的吸光度值差不多,是不是还应该有步骤啊?请专家帮忙指正。
维生素的标准谱图,不知道哪里有???
在检测维生素E和维生素A时,遇到一个棘手的问题。标准品是膏状的,标示为不少于100mg/每瓶,为了配制标准工作液,是直接吸标准品称量配制,还是配制后标定,还是全部配制? 希望有这方面经验的老师,帮忙解答下。
请问测试维生素K1有什么标准?
如题,求液相维生素A标准品的配制方法
谁有维生素A、D、E标准溶液标定特别精准的方法,指点下吧。。。。
[color=#444444]大家好:[/color][color=#444444][/color][color=#444444] 哪位做分析的高手出来分享下用HPLC测定饮料中维生素B族和Vc的标准吧,国内的方法都是比较原始的,想看看欧盟和AOAC的方法,但不太会查找,求帮助!先谢过了[/color]
按国标方法标定维生素A、D、E的标准溶液时,标定结果总是不稳定,即使是新配的标准溶液也一样,大家有没有遇到同样的情况,请高手指点指点!
小白一枚,做维生素C钠和维生素C中的汞时遇到很神奇的事情,首先配制的是汞标,分别为0.5ppb、1ppb、1.5ppb,然后样品浓度为100mg/ml,用标准曲线法测定,线性很好,但是做回收率的时候,回收率能达到300%-400%。然后我又将维生素C消解了一下,重新做回收率,这回回收率达到75%。实在是想不明白为啥,请各位大神指点一下,小弟先在此谢过
维生素A和维生素C的测定:样品处理和提取的区别 首先,我们来看看维生素A。维生素A主要存在于动物性食物中,比如肝脏、鱼肝油、蛋黄等等。在测定维生素A时,样品处理可是个技术活儿。咱们得先把样品捣碎、搅拌均匀,然后加入一些有机溶剂,比如石油醚、乙醚等等,把维生素A从样品中提取出来。这个过程有点像泡茶,只不过咱们泡的是维生素A。 接下来,咱们说说维生素C。维生素C主要存在于水果和蔬菜中,比如橙子、猕猴桃、菠菜等等。维生素C这家伙比较“娇气”,很容易被氧化,所以样品处理时要特别小心。一般来说,咱们得先把样品切碎、榨汁,然后迅速加入一些抗氧化剂,比如草酸、偏磷酸等等,防止维生素C被氧化。这个过程有点像做沙拉,只不过咱们更注重保鲜。 提取方法上,维生素A和维生素C也有区别。维生素A的提取方法主要有皂化法和柱层析法。皂化法就像给维生素A洗个澡,把杂质洗掉;柱层析法就像给维生素A过筛子,把杂质筛掉。而维生素C的提取方法主要有碘量法和2,6-二氯靛酚滴定法。碘量法就像给维生素C称体重,看看它有多少;2,6-二氯靛酚滴定法就像给维生素C量身高,看看它有多高。 通过以上对比,咱们可以看出,维生素A和维生素C在样品处理和提取方法上有着明显的区别。维生素A的样品处理注重提取和净化,而维生素C的样品处理则注重保鲜和抗氧化。在提取方法上,维生素A多采用皂化法和柱层析法,而维生素C则多采用碘量法和2,6-二氯靛酚滴定法。
[color=#444444]饲料中维生素A/E的测定高效液相色谱法,其中第二法维生素A/E乙酸酯和维生素A/E有什么关系啊?怎么换算?要求测定的是维生素A/E,标准品用的是维生素A/E乙酸酯,测定出来的就是维生素A/E乙酸酯吧?还有取用这些标准品该用什么器具呢?[/color]
大家好,我是一名新手,正在做维生素A,D,E,图是跑出来了,但是不知道如何做标准曲线图,知道的说一下,提供一下方式方法,本人学习一下,谢谢,是通过表格还是在仪器软件上做,有做过的可以加微信交流一下:ycxzql1989.谢谢各位朋友了。
如题,是用来做液相的标准曲线的,主要检测饲料里的维生素。有没有价个适中的推荐几个!!谢谢
哪位老师能告诉我维生素E标准品的价格,生产厂家,包装规格,谢谢
求助aocs关于油脂中维生素E的测定标准,谢谢!!
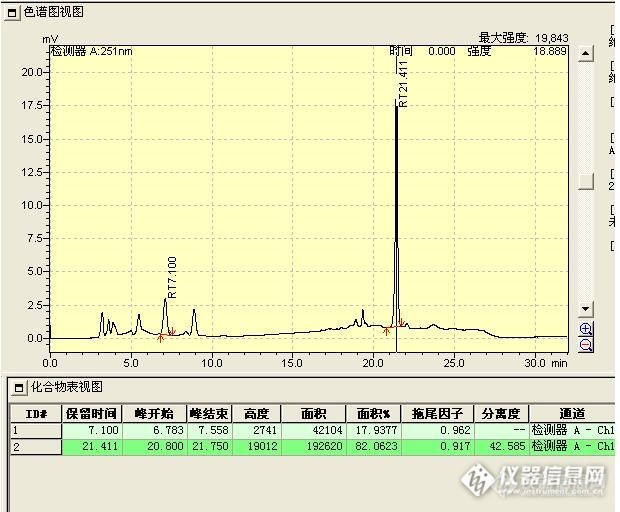
前段时间拿到一个饲料样品,要检测饲料中维生素A和维生素K3,看标准两者用的流动相都是甲醇-水,第一反映就是不是可以梯度洗脱同时测定两个成分,采用定时变换检测波长的方法实现检测。结果很失败,在此分享一下分析过程。 先看标准方法: GB/T 17817-2010 饲料中维生素A的测定 高效液相色谱法[img=,682,136]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292030_01_1638724_3.png[/img][img=,690,336]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292030_02_1638724_3.png[/img] GB/T 7294-2009 饲料添加剂亚硫酸氢钠甲萘醌(维生素K3)[img=,690,506]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292024_01_1638724_3.png[/img] GB/T 18872-2002 饲料中维生素K3的测定 高效液相色谱法 维生素K3的检测原理是在碱性条件下,水溶性的亚硫酸氢钠甲萘醌脱去亚硫酸氢钠生成脂溶性的甲萘醌,用三氯甲烷萃取,吸取适量经甲醇稀释后上机检测。[img=,680,508]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292025_01_1638724_3.png[/img]一、第一次实验:按标准维生素K3的提取过程太麻烦了,且三氯甲烷并不是随便可以买到,那能不能像维生素A一样直接甲醇提取后检测?1、查维生素K3(亚硫酸氢钠甲萘醌)理化特性:易溶于水和热乙醇,难溶于冰乙醇,不溶于苯和乙醚,水溶液PH4.7-7.常温下稳定,遇光易分解。看下面的结构式:[img=,247,199]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292009_01_1638724_3.png[/img]看性质肯定可以溶解在90%甲醇-水里,取亚硫酸氢钠甲萘醌适量,在棕色容量瓶中90%甲醇溶解后上机检测。2、结果出峰极快,受亚硫酸根强极性基团的影响,维生素K3在反相色谱上完全不保留且峰形很差,说明不能直接提取后上机检测,第一次以失败告终。[img]http://ng1.17img.cn/bbsfiles/images/2017/09/201709081927_01_1638724_3.jpg[/img]二、第二次实验:老实的按标准方法(GB/T 18872-2002 饲料中维生素K3的测定 高效液相色谱法)?显然不好,还是不能同时测定维生素A和维生素K3,能不能碱性条件下脱亚硫酸氢钠后纯甲醇提取后上机检测呢?1、由于维生素A的提取方法是65度纯甲醇超声提取30分钟,那么就先取适量维生素A和维生素K3置100mL棕色容量瓶中,由于手上没有氢氧化铵,所以加入5mL碳酸钠溶液代替,加入后超声振摇1min,即有白色沉淀析出(可能是碳酸钠与甲萘醌的混合物),立即加入80mL甲醇,65度超声提取30分钟,过滤取,取滤液稀释至合适浓度上机检测。2、结果几乎无甲萘醌峰出现,观察提取液的颜色,基本可以确定是甲萘醌都氧化成了[color=#cc0000]羟基苯醌[/color]。如下呈褐色的滤液。第二次失败。[img=,602,337]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292059_01_1638724_3.png[/img] 还有一个原因是用甲醇提取与用三氯甲烷提取有明显的区别,是甲醇与碳酸钠溶液是有一定互溶性的,而三氯甲烷与碳酸钠溶液不互溶。甲醇的碳酸钠溶液可能也会促进甲萘醌的破坏。三、第三次实验1、实验方法:维生素A与维生素K3分别按标准方法提取,再吸取甲萘醌的三氯甲烷溶液适量置提取好的维生素A提取液中,混匀,上机检测,梯度洗脱程序如下:[img=,509,384]http://ng1.17img.cn/bbsfiles/images/2017/09/201709292142_01_1638724_3.png[/img]2、结果甲萘醌与维生素A可以实现分离与同时检测,但每针的分析时间太长,与分别单独检测维生素A与维生素K3,每针分析时间只要10分钟相比,没有优势,如果还要同时分析维生素D与维生素E的话,分析时间就会更长,另外也有可能存在提取维生素A时有少量甲萘醌生成,造成结果偏高的误差。其它实验证明维生素A、D、E可能在98%甲醇下同时分析。至此,第三次实验也算失败。[img=,620,512]http://ng1.17img.cn/bbsfiles/images/2017/09/201709300842_01_1638724_3.jpg[/img] 维生素K3(7.1min)与维生素A(21.4min)同时检测图谱[img=,581,500]http://ng1.17img.cn/bbsfiles/images/2017/09/201709300842_02_1638724_3.jpg[/img] 维生素A样品图谱(98%甲醇等度洗脱)[img=,588,492]http://ng1.17img.cn/bbsfiles/images/2017/09/201709300842_03_1638724_3.jpg[/img] 维生素K3样品图谱(80%甲醇等度洗脱) 综上,本次最终还是选择按标准方法分别检测维生素A与维生素K3,在检测维生素K3由标准方法的旋转振荡器改为普通摇床振荡。这次的经验告诉我,要改进方法真的不容易,进行方法开发前一定要充分了解待测组分的理化性质,充分理解参考文献的检测原理与注意事项。看到亚硫酸根有没有人会考虑使用离子对试剂四丁基氢氧化铵和四丁基溴化铵的,估计也是不行的,碱性条件下会水解。
哪里可以买到符合USP标准的维生素C对照品?有USP证书的。
要做维生素A,文献说要甲醇溶解标准品,可惜根本不容,谁做过,帮忙下,怎么溶解。用液相做
急!朋友们!能否提供购买维生素标准品的联系方式.你们用的标准品是进口的呢?还是国产的,二者有没有大的区别!可直在站内给我发消息.也可加我的QQ271017409 谢谢了.
油脂中维生素A和维生素E的测定方法
维生素C的理化性质维生素C又称为抗坏血酸,是含有α-酮基内酯的6个碳原子的酸性多羟基化合物。抗坏血酸为无色无臭的片状结晶体,有酸味、易溶于水、不溶于脂溶剂,遇氧、光、热极易氧化,在碱性环境、加热或与痕量铜、铁等金属离子共存时极易被破坏,在酸性条件下稳定。抗坏血酸C2和C3位两个相邻的烯醇式羟基极易解离、释放出H+,是很强的还原剂。维生素C的生理功能维生素C是一种抗氧化极强的物质,可以有效清除长期暴露在不良环境中所产生的自由基物质。维生素C还有促进胶原形成、矿物质吸收、神经递质的合成、脂肪酸的代谢等作用。维生素C的缺乏与过量人体内缺乏维生素C生物合成必须的葡萄糖酸内酯酶,故体内不能合成维生素C,而必须由膳食供给,否则容易造成维生素C缺乏,导致坏血病。然而,服用过多维生素C可能会出现一些不良反应。维生素C在体内分解代谢最终的重要产物是草酸,长期服用可出现草酸尿以致形成泌尿道结石。维生素C的摄入量根据中国营养学会建议的膳食参考摄入量(RNI):? 成人及孕早期妇女维生素C的推荐摄入量为100mg/d? 中、晚期孕妇及乳母维生素C的推荐摄入量为130mg/d。? 维生素C的可耐受最高摄入量(UL)为1000mg/d[2]我国制定的成年人维生素C的RNI为100mg/d,UL为1000mg/d。在高温、寒冷、缺氧条件下劳动或生活,经常接触铅、苯、汞的有毒作业人群,某些疾病的患者,孕妇、乳母,维生素C的摄入量可适当增加。维生素的主要来源是新鲜的蔬菜和水果。深色蔬菜如冬寒菜、豌豆苗、韭菜、辣椒、油菜苔、花菜、苦瓜等含有丰富的维生素C。水果中以柑、桔、橙、柚、柿、枣和草莓含量丰富,而苹果、梨含量很少;猕猴桃、刺梨、醋柳、酸枣等不仅维生素C含量丰富,而且含有保护维生素C的生物类黄酮。维生素C的检测方法及标准规定目前,蔬菜、水果及其制品中总抗坏血酸的检测方法标准为GB5009.86-2016《食品安全国家标准 食品中抗坏血酸的测定》,包括高效液相色谱法、荧光法和2,6-二氯靛酚滴定法。婴幼儿食品和乳品中维生素C的检测方法则使用GB5413.18-2010《婴幼儿食品和乳品中维生素C的测定》进行检测。另外,GB 14880-2012《食品安全国家标准 食品营养强化剂使用标准》对维生素C在食品中的使用量也做了规定。
维生素C是一种已糖醛基酸,有抗坏血病的作用,所以被人们称做抗坏血酸,主要为还原型及脱氢型两种,广泛存在于植物组织中,新鲜的水果、蔬菜,特别是枣、辣椒、苦瓜、柿子叶、猕猴桃、柑橘等食品中含量较多。它是氧化还原酶之一,本身易被氧化,但在有些条件下又是一种抗氧化剂。维生素C(还原型)纯品为白色无臭结晶,熔点190~192℃,溶于水或乙醇中,不溶于油剂。在水溶液中易被氧化,在碱性条件下易分解,在弱酸条件中较稳定,维生素C开始氧化为脱氢型抗坏血酸(有生理作用)。如果进一步水解则生成2,3-二酮古乐糖酸,失去生理作用。根据它具有的还原性质可以测定维生素C的含量。常用的测定方法有(1)2,6-二氯靛酚法 (还原型VC)(2)2,4-二硝基苯肼法 (总VC)(3)碘酸法(4)碘量法(5)荧光分光光度法一、2,6-二氯靛酚滴定法1、原理:还原型抗坏血酸还原染料2,6-二氯靛酚,该染料在酸性中呈红色,被还原后红色消失。还原型抗坏血酸还原2,6-二氯靛酚后,本身被氧化成脱氢抗坏血酸。在没有杂质干扰时,一定量的样品提取液还原标准2,6-二氯靛酚的量与样品中所含维生素C的量成正比。2、试剂⑴ 1%草酸溶液:称取10g草酸,加水至1000ml;⑵ 2%草酸溶液:称20g草酸,加水至1000ml;⑶ 维生素C标准液:准确称20mgVC溶于1%草酸中,并稀释至100ml,吸5ml于50ml容量瓶中,加入1%草酸至刻度,此溶液每毫升含有0.02mgVC;⑷ 0.02%2,6-二氯靛酚溶液:称取2,6-二氯靛酚50mg,溶于200ml含有52mg碳酸氢钠的热水中,冷却后,稀释至250ml,过滤于棕色瓶中,贮存于冰箱内,应用过程中每星期标定一次。[font=宋
维生素A和维生素D加入无水葡萄糖会造成维生素A和维生素D降解或减小维生素D和维生素A的检出量吗







 我要推广仪器
我要推广仪器
 下载APP
下载APP