灰茅根进入甘肃药典 |多级标准把控我国药品质量
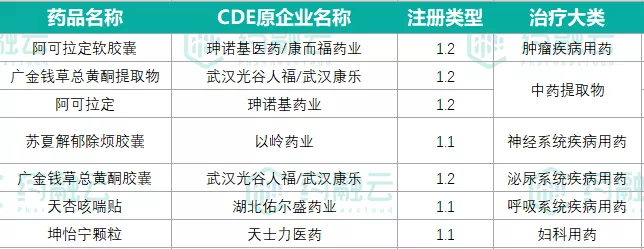
p style=" text-align: justify text-indent: 2em " span style=" color: rgb(0, 112, 192) " strong 灰茅根:不仅具有药用价值 /strong /span br/ /p p style=" text-align: justify text-indent: 2em " 甘肃徽县有一个变异种的狼尾草叫 span style=" color: rgb(255, 0, 0) " strong 灰茅根 /strong /span ,经过6年的实验观察,发现它具有 span style=" color: rgb(149, 55, 52) " strong 消炎、利尿、抗炎、生肌、降血压 /strong /span 等作用。 br/ /p p style=" text-align: justify text-indent: 2em " 近年来,通过徽县人民医院科研团队长期大量走访乡镇卫生院,并寻求多位民间著名中医大夫结合临床中的实际使用,为灰茅根的应用提供共支持。 /p p style=" text-align: justify text-indent: 2em " 根据《中国植物志》、《中国经济植物志》、《陕甘宁青中草药选》等记载,该药材不但药用价值巨大,还可以作为饲料。因为灰茅根富含较高的赖氨酸,可以为当地牧区提供较丰富的料草。另外,还可以编织或造纸、固堤防沙,同时花具有观赏性。 /p p style=" text-align: justify text-indent: 2em margin-top: 10px " span style=" color: rgb(0, 112, 192) " strong 作为中藏药进入甘肃省药典 /strong /span br/ /p p style=" text-align: justify text-indent: 2em " 徽县人民医院联合甘肃省药品检验研究院提出这种变异种狼尾草为一种新中药,上报了省药品监督管理局评审。经过专家组技术评审、国家药品监督管理局审批、公示,顺利通过质量标准认定,进入了甘肃省药典。为彰显徽县特定中药材,正式命名为“灰茅根”。 /p table style=" border-collapse:collapse " width=" 648" align=" center" tbody tr class=" firstRow" td style=" border: 1px solid rgb(255, 255, 255) word-break: break-all " width=" 216" valign=" top" p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202009/uepic/e0b841cb-4b6b-4153-8f42-8e637b3a890c.jpg" title=" 1.png" alt=" 1.png" / /p /td td style=" border: 1px solid rgb(255, 255, 255) word-break: break-all " width=" 216" valign=" top" p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202009/uepic/c3eaf6bf-096b-4de7-a4a7-4721df27acb9.jpg" title=" 2.png" alt=" 2.png" / /p /td td style=" border: 1px solid rgb(255, 255, 255) word-break: break-all " width=" 216" valign=" top" p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202009/uepic/22ccd859-68d4-4f0b-96f0-1f232b9ccfa2.jpg" title=" 3.png" alt=" 3.png" / /p /td /tr /tbody /table p style=" text-align: justify text-indent: 2em " 7月10日,甘肃省药监局发布了一则关于小石韦等25种甘肃省中藏药材标准的公告。参照国家药品标准制定和编写技术要求,根据《省级中药饮片炮制规范修订的技术指导原则》和《甘肃省中藏药材地方标准审定发布工作规范(试行)》, span style=" color: rgb(149, 55, 52) " strong 小石韦、毛细辛、巴夏嘎、牛蒡根、甘肃刺五加、甘肃棘豆、 span style=" color: rgb(255, 0, 0) " 灰茅根 /span 、红药子(撮合散)、李仁、秃疮花、角蒿、苦豆子、油橄榄叶、鬼针草、珠子参叶、盐生肉苁蓉、莳萝子、铁丝威灵仙、铁棒锤、野山楂、硬前胡、菠菜子、椒目、墓头回、缬草 /strong /span strong 25种 /strong 甘肃省中藏药材标准,经省药品检验研究院标准检验复核、省药监局组织专家技术审评、网上公示征求意见,符合甘肃省中藏药材标准要求。 /p p style=" text-align: justify text-indent: 2em margin-top: 10px " strong span style=" color: rgb(0, 112, 192) " 药品标准—还有省药典? /span /strong br/ /p p style=" text-indent: 2em text-align: justify " 此次进入甘肃省药典的25种药材都有了相应的省级标准,在《中国药典》里面没有相应标准的时候需要参考地方药典。 br/ /p p style=" text-indent: 2em text-align: justify " 我国的药品标准有局颁标准、部颁标准、药典,这三者都属于国家标准。现行《中国药典》的权限最高(并不是标准要求最高),以前卫生部颁布的叫部颁标准,成立药监局颁布的叫部颁标准。而企业标准和地方标准只能高于药典,但法律效力不及药典。 br/ /p p style=" text-indent: 2em text-align: justify " 我国药品标准构成如下: /p p style=" text-indent: 2em text-align: justify " 1) 中国药典2020年版、2015年版、2010年版(含勘误)、2005年版、2000年版;中国药典2002、2004年增补本;2005年版勘误; 2006年、2009年增补本;1995年、1990年、1985年、1977年、1963年版。 /p p style=" text-indent: 2em text-align: justify " 2) 卫生部中药成方制剂一至二十册、二十一册(中药保密品种); /p p style=" text-indent: 2em text-align: justify " 3) 卫生部化学、生化、抗生素药品第一分册; /p p style=" text-indent: 2em text-align: justify " 4) 卫生部药品标准(二部)一册至六册; /p p style=" text-indent: 2em text-align: justify " 5) 卫生部药品标准藏药第一册、蒙药分册、维吾尔药分册; /p p style=" text-indent: 2em text-align: justify " 6) 卫生部新药转正标准1至88册; /p p style=" text-indent: 2em text-align: justify " 7) 国家药品标准化学药品地标升国标一至十六册;国家药品标准化学药品地标升国标一至十六册勘误; /p p style=" text-indent: 2em text-align: justify " 8) 国家中成药标准汇编、内科心系、内科肝胆、内科脾胃、内科气血津液、内科肺系、内科肾系、外科妇科、骨伤科、口腔肿瘤儿科、眼科耳鼻喉皮肤科、经络肢体脑系分册; /p p style=" text-indent: 2em text-align: justify " 9) 国家药监局和国家药典委员会颁布的单页标准、新药批件及修订批件; /p p style=" text-indent: 2em text-align: justify " 10) 1999年以来的进口药品标准。 /p p style=" text-indent: 2em text-align: justify " 11)药品检验补充方法与项目(检查特定药品是否造假的检测项目和方法)。 /p p style=" text-indent: 2em text-align: justify margin-top: 10px " span style=" color: rgb(255, 0, 0) " strong 关于2020年版《中国药典》欢迎参加有奖调研: /strong /span br/ /p p style=" text-align:center" a href=" http://instument1999.mikecrm.com/lGWNMkR" target=" _blank" img style=" max-width: 100% max-height: 100% width: 550px height: 179px " src=" https://img1.17img.cn/ui/bimg/SH100000/special/w920h3002020ChP.jpg" title=" " alt=" " width=" 550" vspace=" 0" height=" 179" border=" 0" / /a /p p style=" text-align: justify text-indent: 2em margin-top: 10px " 仪器信息网将特别推出“ span style=" color: rgb(255, 0, 0) " strong 2020年版《中国药典》变化盘点 /strong /span ”专题,盘点通则增修、药典仪器以及相关资讯。敬请广大读者关注! span style=" color: rgb(0, 112, 192) " strong 【点击图片进入专题】 /strong /span /p p style=" text-align: center " img style=" max-width: 100% max-height: 100% width: 550px height: 94px " src=" https://img1.17img.cn/17img/images/202009/uepic/1a99183e-131f-46da-b578-e18bff0eb239.jpg" title=" w640h1102020ChP.jpg" alt=" w640h1102020ChP.jpg" width=" 550" vspace=" 0" height=" 94" border=" 0" / /p

















 我要推广仪器
我要推广仪器
 下载APP
下载APP