各位大哥大姐,能否提供点血球计数仪的资料,谢谢!
论坛中各位大神,我想问问HJ755-2015标准里面用来估算标准菌株个数的血球计数器,一般型号的血球计数器就能够满足要求吗?有些人应该用菌落计数器,还有人说用血球计数板就可以啦!我就想买一台自动估算标准菌株个数的血球计数器,烦请各位大神推荐一下,不甚感激!
我用显微镜观察血球计数板,就是看不到网格线,这是怎么回事?
不知道坛里是否有朋友用过血球计数板计数法测细菌总数的?结果准确吗?
新买的血球计数板到了,可问题却出现了。。。今天上午用显微镜找了半天也没找到计数室有没有朋友用过啊帮忙说说啥原因
血球分析仪哪家的好!(进口的)
由于传统的血球计数板已不能满足高速发展的细胞研究需要,市场上各种自动细胞计数的设备越来越多。常见的主要分为两 类:基于图像的细胞计数仪(Automated vision-based counter)和基于库尔特电阻抗原理的细胞计数仪。两者主要区别在于,前者扫描仪器视野内图像,依靠设定的上下限细胞大小来进行图像识别,而后者根据细胞通过小孔引起电位变化来计算细胞个数(关于库尔特原理,详见此处),两者原理不同,因此所获得的效果也大相径庭。以下对各类细胞计数仪的计数准确性、可重复性、快捷性做结果数据比较,建议读者根据您自身需要选购适合您使用的细胞计数仪。【准确性比较】http://www.bioon.com/tech/UploadFiles_3081/201107/2011072416480295.jpg图1. 不同细胞浓度下各种细胞计数仪的计数结果与实际数值的对比上图可见:血球计数板在高细胞密度 时,计数结果与实际细胞密度有偏差,而基于图像的细胞计数仪则在多个浓度下均有较大偏差。这表明基于库尔特原理的细胞计数仪(Coulter counter和Scepter cytometer)在计数准确性上优于其他两种细胞仪。(样品为常见的COS7细胞)【可重复性比较】http://www.bioon.com/tech/UploadFiles_3081/201107/2011072416514090.jpg 图3. 不同细胞浓度下细胞计数的可重复性比较上图可见:基于库尔特原理细胞计数仪的可重复性均明显好于血球计数板和基于图像的细胞计数仪。(样品为19种细胞系)【快捷性】http://www.bioon.com/tech/UploadFiles_3081/201107/2011072416541552.jpg图5. 三种细胞计数法的计数时间对比上图可见:传统的血球计数板的计数时间远长于其他两种计数。库尔特原理计数法与基于图像的细胞计数法相比,计数速度更为稳定,而且计数时间约为后者的一半。(样品为常见的SF9,MCF7,HEK293细胞)
分享饲料用喷雾干燥血球粉的国家标准
哪里有的买,我要厂家的,不要代理的。我急啊?要电话和网站
蔡康显微镜下的观察纤维细胞的微管分布-技术精华 目前显微镜观察微生物有:生物显微镜 蔡康高档显微镜 荧光显微镜 倒置显微镜 都可以观察研究微生物,请看下面阐述介绍 [img=340,340]http://www.zskp.org.cn/Article/UploadFiles/200803/2008030615471734.jpg[/img]一、目的要求 1.明确显微镜计数的原理。 2.学习使用血球计数板进行微生物计数的方法。 二、基本原理 利用血球计数板在显微镜下直接计数,是一种常用的微生物计数方法。此法的优点是直观、快速。将经过适当稀释的菌悬液(或孢子悬液)放在血球计数板载玻片与盖玻片之间的计数室中,在显微镜下进行计数。由于计数室的容积是一定的( 0.1mm2),所以可以根据在显微镜下观察到的微生物数目来换算成单位体积内的微生物总数目。由于此法计得的是活菌体和死菌体的总和,故又称为总菌计数法。 血球计数板,通常是一块特制的载玻片,其上由四条槽构成三个平台。中间的平台又被一短横槽隔成两半,每一边的平台上各刻有一个方格网,每个方格网共分九个大方格,中间的大方格即为计数室,微生物的计数就在计数室中进行。血球计数板构造如图Ⅷ-1。 计数室的刻度一般有两种规格,一种是一个大方格分成16个中方格,而每个中方格又分成25个小方格(图Ⅷ-2);另一种是一个大方格分成25个中方格,而每个中方格又分成16个小方格(图Ⅷ-1,C)。但无论是哪种规格的计数板,每一个大方格中的小方格数都是相同的,即16×25=400小方格,如图Ⅷ-2。 每一个大方格边长为1mm,则每一大方格的面积为1mm2,盖上盖玻片后,载玻片与盖玻片之间的高度为0.1mm,所以计数室的容积为0.1mm3。 在计数时,通常数五个中方格的总菌数,然后求得每个中方格的平均值,再乘上16或25,就得出一个大方格中的总菌数,然后再换算成1ml菌液中的总菌数。 下面以一个大方格有25个中方格的计数板为例进行计算:设五个中方格中总菌数为A,菌液稀释倍数为B,那么,一个大方格中的总菌数 因1ml=1cm3=1000mm3, (即0.1mm3中的总菌数)为(A/5)*25*B=50000AB(个) 同理,如果是16个中方格的计数板,设五个中方格的总菌数为A',则 1ml菌液中总菌数=(A'/5)*16*10*1000*B'=32000A'B'(个) 三、器材 酿酒酵母菌悬液,血球计数板,显微镜,盖玻片,无菌毛细管。 四、操作步骤 1.稀释 将酿酒酵母菌悬液进行适当稀释,菌液如不浓,可不必稀释。 2.镜检计数室 在加样前,先对计数板的计数室进行镜检。若有污物,则需清洗后才能进行计数。 3.加样品 将清洁干燥的血球计数板盖上盖玻片,再用无菌的细口滴管将稀释的酿酒酵母菌液由盖玻片边缘滴一小滴(不宜过多),让菌液沿缝隙靠毛细渗透作用自行进入计数室,一般计数室均能充满菌液。注意不可有气泡产生。 4.显微镜计数 静止5分钟后,将血球计数板置于显微镜载物台上,先用低倍镜找到计数室所在位置,然后换成高倍镜进行计数。在计数前若发现菌液太浓或太稀,需重新调节稀释度后再计数。一般样品稀释度要求每小格内约有5—10个菌体为宜。每个计数室选5个中格(可选4个角和中央的中格)中的菌体进行计数。位于格线上的菌体一般只数上方和右边线上的。如遇酵母出芽,芽体大小达到母细胞的一半时,即作两个菌体计数。计数一个样品要从两个计数室中计得的值来计算样品的含菌量。 5.清洗血球计数板 使用完毕后,将血球计数板在水笼头上用水柱冲洗,切勿用硬物洗刷,洗完后自行晾干或用吹风机吹干。镜检,观察每小格内是否有残留菌体或其他沉淀物。若不干净,则必须重复洗涤至干净为止。 五、实验报告 1.结果 将结果记录于下表中。 A表示五个中方格中的总菌数; B表示菌液稀释倍数。 2.思考题 根据你实验的体会,说明用血球计数板计数的误差主要来自哪些方面?应如何尽量减少误差,力求准确?这是多年以来上海蔡康光学经历磨炼 得出的结果,也为研究人员带来的方便,为国家作出贡献.
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=174487]GB 23875-2009 饲料用喷雾干燥血球粉[/url]
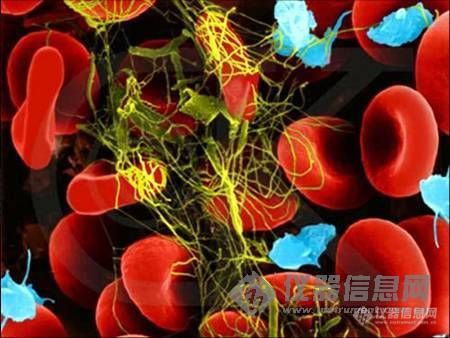
扫描电镜下血液凝块构造(红色为红血球,兰色为血小板,黄色为纤维蛋白)。[img]http://ng1.17img.cn/bbsfiles/images/2005/12/200512071347_11393_1697463_3.jpg[/img][color=blue]Sean:非常感谢楼主一直以来的好图,同时也希望大家踊跃将自己的图片发上来跟大家分享、讨论!![/color]
离心PBS液1 ml洗涤,吹打制成待测细胞悬液取1 ul悬液+9 ul PBS细胞计数:1. 盖好盖玻片:取一套血球计数板,将特制的盖玻片盖在血球计数槽上.2. 用10 ul枪头将细胞悬液注入计数板,要保证盖片下充满悬液,注意盖片下不要有气泡,也不能让悬液流入旁边槽中。3. 统计四个大格的细胞数:将血球计数板放于显微镜的低倍镜下观察,并移动计数板,当看到镜中出现计数方格后,数出四角的四个大铬(每个大格含有16个中铬)的细胞数目。4. 计算原细胞悬液的细胞数:按照下面公式计算细胞密度: (细胞悬液的细胞数)/ml=(四个大格子细胞数/4)×106
[URL=http://jiangsu.chinainfoseek.com/]江苏招标网[/URL]受常州市戚墅堰区丁堰街道社区卫生服务中心的委托,现就三分类血球分析仪进行采购,有关事项公告如下:一、投标人必须符合的条件:1、国内注册生产或经营本次采购设备、注册资本在50万元(含)以上具有独立法人资格、有履行合同所必需的设备和专业技术能力的企业;2、在以前采购活动中没有违法和不良记录;3、具有医疗器械经营企业许可证,投标产品具有相应的医疗器械注册证;4、持有制造商(或具有同等法律效力的授权机构)的代理授权书;二、资格审查、报名时间:时间:2009年11月27日起至2009年12月4日正常工作时间在该期限后提出的申请将不再受理。地点:常州市政府采购中心二楼采购一科。三、报名时须提交的文件资料①身份证(原件及复印件),②企业营业执照副本(原件及复印件加盖公章),③法定代表人授权委托书(原件)。④医疗器械经营企业许可证(原件及复印件加盖公章)⑤投标产品相应的医疗器械注册证,符合中国政府规定的相应技术、安全和环保标准(复印件加盖公章)⑥持有制造商(或具有同等法律效力的授权机构)的代理授权书(原件及复印件加盖公章)注:报名单位如未在中心办理供应商登记需要通过资格审查并登记备案。四:联系方式:联系人:李新彦电话:0519-86800187地点:常州市晋陵中路593号信息来源:[URL=http://www.chinainfoseek.com/]中国采购与招标网[/URL],中国招标采购导航网http://www.chinainfoseek.com/
微生物细胞大小的测定和显微镜直接计数 目的 1.1 学习接目测微计的校正方法, 了解血球计数板的构造和计数原理 1.2 学习使用显微镜测微尺测定微生物细胞大小, 掌握用血球计数板测定微生物细胞总数 的方法。 2 原理 微生物细胞的大小是微生物分类鉴定的重要依据之一。微生物个体微小,必须借助于显微镜才能观察,要测量微生物细胞大小,也必须借助于特殊的测微计在显微镜下进行测量。 显微测微计由镜台测微计和目镜测微计两部分组成。后者可直接用于测量细胞大小。它是一块圆形玻片(图7—1),其中央有精确等分到度,测量时将其放在接目镜中的隔板上。由于目镜测微计所测量的是微生物细胞经过显微镜放大之后所成像的大小,刻度实际代表的长度随使用的目镜和物镜放大倍数及镜筒的长度而改变,所以,使用前须先用镜台测微计进行标定,求出某一放大率下,目镜测微计每一小格所代表的长度,然后用目镜测微计直接测被测对象的大小。镜台测微计是一块中央有精确刻玻片(图7—1),刻度的总长为lmm,等分为100小格,每小格长10um,专用于对目镜测微计进行标定的。 3 材料 3.1 器械 显微镜、目镜测微尺、镜台测微尺,载玻片、盖玻片、血球计算板、擦镜纸、吸水纸、玻片架、肾形盘、洗瓶、接种环、酒精灯、火柴、滴管。 3.2 菌种 培养48h的啤酒酵母斜面菌体和菌悬液。 3.3 革兰氏染液 4流程 4.1 置目测微计→置台测微计→标定目测微计→测菌体大小→记录结果→用毕擦拭干净 4.2 检查计数板→稀释样品→加样→计数→计算→清洗 5 步骤 5.1 微生物菌体大小的测定 5.1.1 目镜测微尺的校正 5.1.1.1 更换目镜镜头 更换目镜测微尺镜头(标记为PF);或者取下目镜上部或下部的透镜,在光圈的位置上安上目镜测微尺,刻度朝下,再装上透镜,制成一个目镜测微尺的镜头。 5.1.1.2 某一倍率下标定目镜刻度 将镜台测微尺置于载物台上,使刻度面朝上,先用低倍镜对准焦距、看清镜台测微尺的刻度后,转动目镜,使目镜测微尺与镜台测微尺的刻度平行,移动推动器使两尺重叠,并使二尺的左边的某一刻度相重合,向右寻找另外二尺相重合的刻度。记录两重叠刻度间的目镜测微尺的格数和镜台测微尺的格数(图7-1C)。 5.1.1.3 计算该倍率下目镜刻度 目镜测微尺每格长度=镜台测微尺格数/目镜测微尺格数xl0um 5.1.1.4 标定并计算其他放大倍率下的目镜刻度 以同样方法分别在不同倍率的物镜下测定目镜测微尺每格代表的实际长度。如此测定后的测微尺的长度,仅适用于测定时使用的显微镜以及该目镜与物镜的放大倍率。 5.1.2 菌体大小的测定 5.1.2.1 将啤酒酵母制成水浸片。 5.1.2.2 大小换算 将标本先在低倍镜下找到目的物,然后在高倍镜下用目镜测微尺测定每个菌体长度和宽度所占的刻度,即可换算成菌体的长和宽。 5.1.2.3 求平均值 一般测量微生物细胞的大小,用同一放大倍数在同一标本上任意测定l0一20个菌体后,求出其平均值即可代表该菌的大小。 5.2 用血球计数板测定微生物细胞的数量 5.2.1 检查血球计数板 取血球计数板一块,先用显微镜检查计数板的计数室,看其是否沾有杂质或干涸着的菌体,若有污物则通过擦洗、冲洗,使其清洁。镜检清洗后的计数板,直至计数室无污物时才可使用。 5.2.2 稀释样品 将培养后的酵母培养液振荡振摇混匀,然后作一定倍数的稀释。稀释度选择以小方格中的分布的菌体清晰可数为宜。一般以每小格内含4~5个菌体的稀释度为宜。 5.2.3 加样 取出一块干净盖玻片盖在计数板中央。用滴管取1滴菌稀释悬液注入盖玻片边缘,让菌液自行渗入,若菌液太多可用吸水纸吸去。静置5—10分钟。 5.2.4 镜检 待细胞不动后进行镜检计数。先用低倍镜找到计数室方格后,再用高倍镜测数。一般应取上下及中央五个中格的总菌数。计数时若遇到位于线上的菌体,一般只计数格上方(下方)及右方(左方)线上的菌体。每个样品重复3次。 5.2.5 计算 取以上计数的平均值,按下列公式计算出每毫升菌液中的含菌量。 菌体细胞数(cfu/mL)=小格内平均菌体细胞数%26#215 400%26#215 104%26#215 稀释倍数 5.2.6 清洗 计数板用毕后先用95%的形酒精轻轻擦洗,再用蒸馏水淋洗,然后吸干,最后用擦镜纸揩干净。若计数的样品是病原微生物,则须先浸泡在5%石炭酸溶被中进行消毒,然后再行清洗。清洗后放回原位,切勿用硬物洗刷。 图7-2 血细胞计数板 6 结果 6.1计算出目镜测微尺在低、高倍镜下的刻度值。 6.2 记录菌体大小的测定结果。 6.3 计算样品中酵母菌浓度。 7 思考 7.1 为什么随着显微镜放大倍数的改变,目镜测微计每格相对的长度也会改变?能找出这种变化的规律吗? 7.2 根据测量结果,为什么同种酵母菌的菌体大小不完全相同? 7.3 能否用血球计数板在油镜下进行计数?为什么? 7.4 根据自己体会,说明血球计数板计数的误差主要来自那些方面?如何减少误差? 8 附录 目镜测数尺有两种:一是特制的目镜镜头,镜片上刻有50等分或100等分的刻度,使用时直接安装在显微镜上,取代没有刻度的目镜镜头;另一种是一块直径大约17.5 mm的圆玻璃片,其中央刻有50等分或100等分的刻度(图7-1A),使用时将该玻璃片安装在原来的目镜镜头上即可。由于不同的显微镜放大倍数不同,既使同一显微镜在不同的目镜、物镜组合下其放大倍数也不同,故目镜测微尺每格实际表示的长度随显微镜放大倍数不同而异。也就是说,目镜测微尺上的刻度只代表相对的长度。因此在使用前须用镜台测微尺校正,以确定在一定放大倍数下目镜测微尺的每格长度。 血球计数板是一块特别的厚玻片,玻片中央分剖成两个平面,上面各刻有9区,中央一区为计数室,供计数用(图7-2A),此区的长和宽各为1mm。中央平面两侧有小沟,小沟外有两条突起的平台,平台比中央平面高0.1mm,因此计数室体积为0.1mm3,容积为10-4ml。通常计数室分为25个大格,每大格又分为16个小格,每小格容积为4%26#215 10-6ml,即lml菌液容积相当于400万个小格体积。因此只要将细胞悬液注人计算室,计算出一定数量小格的平均菌数即可算出每毫升的细胞数。
免疫共沉淀技术原理
[font=&]【题名】: 现代仪器分析原理与技术 [/font][font=&]【全文链接】: https://mtoou.info/jueban/750484.html[/font]
浊度仪的原理与技术方案 浊度仪(浊度计)一浊度计/浊度仪原理浊度是表现水中悬浮物对光线透过时所发生的阻碍程度。水中含有泥土、粉尘、微细有机物、浮游动物和其他微生物等悬浮物和胶体物都可使水中呈现浊度。本浊度仪(浊度计)采用900散射光原理。由光源发出的平行光束通过溶液时,一部分被吸收和散射,另一部分透过溶液。与入射光成900方向的散射光强度符合雷莱公式:Is=((KNV2)/λ)×I0其中:I0——入射光强度Is——散射光强度N——单位溶液微粒数V——微粒体积λ——入射光波长K——系数在入射光恒定条件下,在一定浊度范围内,散射光强度与溶液的混浊度成正比。上式可表示为:Is/I0= K′N(K′为常数)根据这一公式,可以通过测量水样中微粒的散射光强度来测量水样的浊度。 浊度仪的原理与技术方案 一、浊度计/浊度仪主要技术性能浊度仪(浊度计)是高精度测量仪,采用四位LED数字显示,具有自动切换量程,稳定准确,使用方便等特点,广泛适用于食品、石油、化工、环境监测、医药卫生等行业。1、测量范围:0~1000度。分0~5、5~25、25~100、100~400、400~800、800-1000六个量程(度是1个Formazine浊度单位,对于散射式仪器,即1NTU)。2、精确度:≤±2 % (满量程) 3、重现性:≤±2 % (满量程) 4、分辨率:0.01 NTU 5、每小时漂移:0.1 NTU6、外形尺寸:282mm×237mm×102mm 7、重量:2.2kg8、仪器在开机通电半小时后可在下列环境下连续运行:⑴环境温度: 5~40℃⑵相对湿度:≤70%⑶供电电源: AC(220±10%)V;50Hz⑷避免强光直接照射,无显著的振动及强电磁干扰
计数器的原理是什么?
游戏操作: 点击“start”开始游戏。 游戏开始以后,小怪物会踩着雪球,沿着山坡慢慢向下滚动。当然,由于受到惯性的作用,雪球滚动的速度会越来越快,而雪球的体积同时也会越来越大。我们可怜的企鹅这回又要倒霉了,看到雪球滚下也不会躲开,玩家所要做得,就是尽可能多的压到企鹅。 想要压到更多的企鹅,当然需要走更长的路。在滚下的过程中,会有一面面的旗帜,标示出地面的裂缝,如果玩家不幸碰上的话,雪球就会停止滚动,所以需要避开它们。方法是按下鼠标左键,让雪球跳起,越过障碍。这里需要提醒的是,由于游戏开始时,雪球的速度比较慢,所以玩家可以在它滚到旗帜的前方才按下跳跃键,而到后期,雪球速度飞快时,只要一看到有旗帜出现,就得立即按下鼠标左键跳跃才行,各位得留神啊!画面下方的数字表示玩家目前通过的距离![Flash]http://image2.sina.com.cn/dongman/f/2004-04-20/U54P55T4D18810F248DT20040420115548.swf[/Flash][Url=http://image2.sina.com.cn/dongman/f/2004-04-20/U54P55T4D18810F248DT20040420115548.swf]右键另存下载[/Url]
[font=&]【题名】:电分析化学原理及仪器使用技术[/font][font=&]【全文链接】: https://mtoou.info/jueban/538463.html[/font]
ICP-MS是上世纪八十年代兴起的测试分析技术,其以谱线简单,干扰少,低背景、低检测限、多元素同时测试、线性动态范围宽等优势成为无机元素分析的佼佼者,是分析技术的一次革命,虽然简单,但能够用的得心应手并非易事,了解一台仪器要从他的组成原理入手,因此请大家多多发表一些与ICP-MS基本结构与基本原理方面的资料,一起来庖丁解牛吧!
想请问专家一个问题,Peltier温控技术的原理以及与传统温控技术相比有哪些优点。谢谢!
便携式气质联用仪有什么特点,技术和原理上和常规的有什么区别
近年来随着生物技术的不断发展,出现了许多克隆新基因的方法和手段,如图谱克隆技术、转座子标签技术、mRNA差异显示技术二基因组减法技术以及cDNA文库筛选技术等。但上述方法人多具有实验周期长、技术步骤烦琐且工作量大等特点。cDNA末端快速扩增技术(rapid amplification of cDNA ends,RACE)是一种基于PCR从低丰度的转录本中快速扩增cDNA的5'和3'末端的有效方法,以其简单、快速、廉价等优势而受到越来越多的重视。经典的RACE技术是由Frohman等(1988)发明的一项技术,主要通过RT-PCR技术由已知部分cDNA序列来得到完整的cDNA5'和3'端,包括单边PCR和锚定PCR。该技术提出以来经过不断发展和完善,克服了早期技术步骤多、时间长、特异性差的缺点(Frohman等,1995:Schaefer,l995: Chen,1998: Bespalova等,1998: Matz等11999)。对传统RACE技术的改进主要是引物设计及RT-PCR技术的改进:改进之一是利用锁定引物((lock docking primer)合成第一链cDNA,即在oligo(dT)引物的3' 端引入两个简并的核苷酸【5'-Oligo(dT)16-30MN-3',M=A/G/C;N=A/G/C/T】,使引物定位在poly(A)尾的起始点,从而消除了在合成第一条cDNA链时oligo(dT)与poly(A)尾的任何部位的结合所带来的影响;改进之二是在5' 端加尾时,采用poly(C),而不是poly(A);改进之三是采用RNase H-莫洛尼氏鼠白血。病毒(MMLV)反转录酶或选择嗜热DNA聚合酶可能在高温(60℃ -70℃)有效地逆转录mRNA,从而消除了5'端由于高CC含量导致的mRNA 二级结构对逆转录的影响;改进之四是采用热启动PCR (hot start PCR)技术和降落PCR(touch down PCR)提高PCR反应的特异性。随着RACE技术日益完善,目前己有商业化RACE技术产品推出,如CLONTECH的MarathoTM技术和SMARTTM RACE技术。邢桂春等(2001)先后使用上述两种试剂盒进行RACE反应,结果发现使用MarathonTM所得到的片断总是比采用SMARTTM RACE试剂盒到所得到的片断短。其原因在于MarathonTM技术反转录反应往往不能真正达到mRNA的5' 末端。所以认为,进行RACE反应应当优选SMARTTM RACE试剂盒。以下就国内目前应用最广的SMARTTM RACE试剂盒为例,简要概述RACE技术的原理和操作过程。SMARTTM 3'-RACE的原理利用mRNA的3'末端的poly(A)尾巴作为一个引物结合位点,以连有SMART寡核营酸序列通用接头引物的Oligo(dT)30MN作为锁定引物反转录合成标准第一链cDNA.然后用一个基因特异引物GSP1(gene specific primer,GSP)作为上游引物,用一个含有部分接头序列的通用引物UPM(universal primer,UPM)作为下游引物,以cDNA第一链为模板,进行PCR循环,把目的基因3' 末端的DNA片段扩增出来。(见Figure 1)http://tong.dxy.cn/upload/asset/2009/10/23/1256188568.jpgFigure 1.SMARTTM 5'-RACE的原理先利用mRNA的3'末端的poly(A)尾巴作为一个引物结合位点,以Oligo(dT)30MN作为锁定引物在反转录酶MMLV作用下,反转录合成标准第一链cDNA.利用该反转录酶具有的末端转移酶活性,在反转录达到第一链的5'末端时自动加上3-5个(dC)残基,退火后(dC)残基与含有SMART寡核苷酸序列Oliogo(dG)通用接头引物配对后,转换为以SMART序列为模板继续延伸而连上通用接头(见Figure 2 )。然后用一个含有部分接头序列的通用引物UPM(universal primer,UPM)作为上游引物,用一个基因特异引物2(GSP 2 genespecific primer,GSP)作为下游引物,以SMART第一链cDNA为模板,进行PCR循环,把目的基因5'末端的cDNA片段扩增出来。最终,从2个有相互重叠序列的3'/ 5'-RACE产物中获得全长cDNA,或者通过分析RACE产物的3'和5'端序列,合成相应引物扩增出全长cDNA。http://tong.dxy.cn/upload/asset/2009/10/23/1256188569.jpgFigure 2.实验中发现,做RACE反应实验实际操作中仍存在不少困难。因此,对RACE反应条件进行反复摸索是十分必要的。这是因为:第一,在5'-RACE包含了有3个连续的酶反应步骤(反转录、同聚物加尾和PCR扩增),每一步都可能导致失败;第二,扩增DNA末端的特异性完全依赖锚定引物及扩增DNA模板样品的不均一性,因而特异性一般很低,常呈现不清晰的成片条带或截短的产物背景。因此,使用RACE技术扩增得到的特异末端片段,所获得的重组克隆最好能够全部测序,以排除RACE实验结果中扩增产物假阳性和假阴性,最终有可能获得新基因的全序列。随着RACE的不断改进和完善,优化条件下PCR扩增效率和忠实性的提高及PCR产物克隆技术的迅速发展,RACE必将在基因克隆及基因表达研究中发挥越来越大的作用。RACE的另一种代表方法-Self-Ligation法1. 反转录(RT)反应。2. Hybrid RNA的分解。3. 单链cDNA的自身连接。4. PCR扩增5'未知区域。5. 目的DNA片段的切胶回收。6. DNA序列测定。原理见Figure 3。http://tong.dxy.cn/upload/asset/2009/10/23/1256188570.jpgFigure 3.
一份不错的PPT,与大家分享,气质联用检测技术原理及应用
耶拿[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]基本原理及其分析技术[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=187512][url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]基本原理及其分析技术.pdf[/url]
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]基本原理与分析技术[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=177760][url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]基本原理与分析技术.pdf[/url]
[font=mp-quote, -apple-system-font, BlinkMacSystemFont, &][b][b][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]一、概念[/font][/b][/b][/font] [b]1.诱变[font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]:[/font][/b] [font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]通过人为的措施诱导遗传物质产生突变的方法。简单来说,诱变就是一种使遗传物质改变的手段。[/font] 目前,主要有三种方法:紫外诱变、化学诱变和等离子体诱变。 [b][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]2.育种:[/font][/b] [font=mp-quote, -apple-system-font, BlinkMacSystemFont, &][size=17px]通过创造遗传变异、改良遗传特性,以培育优良新品种的技术。育种的目的就是改良新品种,挑选优良品种,让其朝着有利人类的方向发展。[/size][/font] [b][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]3.微生物诱变育种:[/font][/b] [font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]以人工诱变手段诱发微生物遗传物质发生突变,并从突变群体中筛选出所需要的突变株,进而培育成新品种的育种方法。[/font] [font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]在这里,作用对象为各类微生物,包括细菌、放线菌、酵母和霉菌等。[/font] [font=mp-quote, -apple-system-font, BlinkMacSystemFont, &][b][b][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]二、诱变菌悬液的标准[/font][/b][/b][/font] [b]1.[font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]菌悬液[/font][/b]将菌种接种至合适的培养基中,培养至[b]对数生长期[/b],离心收集菌体,用无菌生理盐水洗涤两次,再用适量生理盐水重悬使菌体浓度调整到1×10[font=&][sup]6[/sup][/font]CFU/mL-1×10[font=&][sup]8[/sup][/font]CFU/mL。[b](1)诱变育种使用对数期菌种的原因[/b]在对数期阶段,细菌生理状态最佳,生长迅速、代谢活跃、对诱变剂的反应敏感,使得细菌在诱变处理时更容易产生遗传变异,诱变效率高,从而增加选育出优良菌株的机会。[b](2)对数期的确定[/b]试验前,绘制生长曲线,确定对数期范围。 试验时,控制培养条件(培养基、温度、pH值等),尽量与制作生长曲线条件一致。 如果没有做生长曲线,怎么办?试验时,定期测定菌悬液中的活菌数即可判断细菌是否处于对数生长期。比如说采用血球计数板计数。 [b]2.孢子悬浮液[/b] 对于产孢子微生物,将菌种接种至固体平板或斜面上培养至产生[b]大量孢子[/b],用生理盐水洗脱并离心收集。用生理盐水洗涤两次,再用适量生理盐水重悬并通过机械振荡使其充分分散,过滤除掉菌丝体。最后用生理盐水悬浮得到均匀的孢子悬浮液,并将其浓度调整到1×10[font=&][sup]6[/sup][/font]CFU/mL-1×10[font=&][sup]8[/sup][/font]CFU/mL。 [b][b][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]三、诱变育种方法[/font][/b] 1.紫外诱变原理:[size=17px][/size][/b][size=17px]通过紫外线照射使DNA分子内形成嘧啶二聚体,造成DNA复制错误或阻碍碱基间的正常配对,从而引起基因突变。紫外诱变使用安全,但已发生突变的细胞遇见可见光会复活,且存活率较高。 [/size] [b](1)紫外诱变波长[/b] 紫外诱变的波长λ一般设定为260nm,实际上波长是253.7nm,在此波长下DNA有最大吸收峰,能够最大程度地诱发DNA结构变化,从而导致基因突变。而同时,[size=17px]蛋白质的吸收峰位于280nm,又不影响蛋白质,这样可以减少对蛋白分子的干扰。[/size] [b](2)紫外照射距离和功率[/b] 在诱变前,需检查紫外灯,并提前预热,保证照射过程中频率稳定,诱变效果稳定。 [size=17px]诱变时,要摇动培养皿中菌种,提高菌种诱变效率,也要控制紫外照射距离和照射时间。 [/size] 紫外线灯的强度随着照射距离的减小而增大。当距离过近时,紫外线强度过高,可能导致微生物细胞在短时间内受到大量紫外线的照射,引起过度的DNA损伤。 [b](3)致死率[/b]不同的菌种,紫外诱变致死率不同,确定致死率,可以确定诱变条件是否合适。紫外照射时间太短,突变率低,达不到预期效果;而照射时间太长,菌体有可能全部死亡。因此,要想保证高的突变率,确定致死率至关重要。研究发现,当致死率在85%左右时,诱变效果最好,正突变率最高。 [b](4)注意事项[/b] 紫外诱变时,一定要注意个人安全,规范佩戴好防护眼镜、防护手套和防护服等。 诱变完成,要注意避光,避免光复活。 2.[b]化学诱变原理:[/b][size=17px]利用化学诱变剂直接改变DNA的结构,造成DNA复制时的碱基配对错误,进而引起基因突变。[/size] [b](1)常用化学诱变剂 (2)化学诱变效率[/b] [size=17px]突变率较高,以点突变为主,具有专一性,而对其余部位无影响。但诱变剂毒性较大,需要严格控制化学诱变剂的浓度和作用时间。 [/size][b](3)注意事项[/b]一定要注意个人安全,规范佩戴好防护眼镜、防护手套和防护服等。 [size=17px]化学诱变通常不具有光复活现象,无需[/size]避光。 [b]3.常压室温等离子体诱变(ARTP) 原理:[/b][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]一种新型高效的生物育种诱变方法,能够在大气压下产生温度在25-40°C之间具有高活性粒子浓度的等离子体射流。[/font][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &]这些高活性粒子(如激发态的氦原子、氧原子、OH自由基等)可以对微生物细胞产生多重作用,如造成遗传物质的损伤、引起细胞膜通透性和蛋白结构的改变等。[/font] [b](1)[size=17px][font=system-ui, -apple-system, BlinkMacSystemFont, &]常压室温等离子(ARTP)诱变育种仪[/font][/size][/b][size=17px][/size] [align=center] [/align][b](2)致死率(3)优势与劣势 [size=17px]优势:[/size][/b][size=17px][/size][size=17px]突变率高,诱变速度快; [/size][size=17px]ARTP设备简单、操作简易、运行成本低廉;[/size][size=17px]ARTP本身对环境无污染和危害。[/size][b]劣势:[/b] [size=17px]设备成本较高;[/size] [size=17px]需要专业的维护和保养。[/size] [b][size=17px](4)注意事项[/size][/b][size=17px] [/size] [size=17px]严格遵守“先开电再开气,先关气再关电”的操作顺序。[/size] [size=17px]需要经过培训后操作。 [/size]
液氮槽vaporphase长期储存。-20℃不可超过1小时,以防止胞内冰晶过大,造成细胞大量死亡,亦可跳过此步骤直接放入-80℃冰箱中,惟存活率稍微降低一些。(2)程序降温:利用已设定程序的等速降温机以-1~-3℃/分钟之速度由室温降至(-80℃以下)-120℃,再放在液氮槽vaporphase长期储存。适用于悬浮型细胞与hybridoma之保存。3、步骤:(1)冷冻前24-48小时更换半量或全量培养基,使细胞处于指数生长期。(2)配制冷冻保存溶液(使用前配制):另取一离心管,加入培养基、血清,逐滴加入二甲基亚砜(DMSO)至20%浓度,即制成双倍的冻存液,置于室温下待用。(3)离心收集培养之细胞,用加血清的培养基重悬起细胞,取少量细胞悬浮液(约0.1ml)计数细胞浓度及冻前存活率。(4)取与细胞悬液等量的冻存液,缓慢逐滴加入细胞悬液,并晃动试管,制成细胞冻存悬液(DMSO最后浓度为5~10%),使细胞浓度为1~5×106cells/ml,混合均匀,分装于已标示完全之冷冻保存管中,1~2ml/vial,并取少量细胞悬浮液作污染检测。严密封口后,注明细胞名称、代数、日期。然后进行冻存。4、注意事项:(1)欲冷冻保存之细胞应在生长良好(logphase)且存活率高之状态,约为80~90%致密度。冷冻前检测细胞是否仍保有其特有性质,例如hybridoma应在冷冻保存前一至二日测试是否有抗体之产生。(2)细胞在液氮中可长期冻存无限时间,而不会影响细胞活力;在-70度可保存数月。(3)注意冷冻保护剂之品质。DMSO应为试剂级等级,无菌且无色(以0.22micron FGLP Telflon过滤或是直接购买无菌产品,如Sigma D-2650),以5~10ml小体积分装,4℃避光保存,勿作多次解冻。Glycerol亦应为试剂级等级,以高压蒸汽灭菌后避光保存。在开启后一年内使用,因长期储存后对细胞会有毒性。本方法中先制备双倍冻存液,可避免DMSO直接加入时释放的热量对细胞的损伤。缓慢逐滴加入细胞悬液是使细胞逐步适应高渗,可降低细胞受损。DMSO可能引起部分白血病细胞株的分化,可换用10%甘油冻存。(4)冷冻保存之细胞浓度:①normal human fibroblast:1~3×106cells/ml②hybridoma:1~3×106cells/ml,细胞浓度不要太高,某些hybridoma会因冷冻浓度太高而在解冻24小时后死去。③adherent tumor lines:5~7×106,依细胞种类而异。Adenocarcinoma解冻后须较高之浓度,而HeLa只需1~3×106cells/ml④other suspensions:5~10×106cells/ml,human lymphocyte须至少5×106cells/ml。(5)冷冻保护剂浓度为5或10%DMSO,若是不确定细胞之冷冻条件,在做冷冻保存之同时,亦应作一个backup culture,以防止冷冻失败。(6)冻存可用10%~90%的血清,一般高浓度血清有助于维护细胞活力,此处介绍20%终浓度有利于细胞悬浮而少沉积(4度时),复苏存活率在80%~90%以上,对原代培养细胞,以90%血清冻存更为有效。二、冷冻细胞活化1、冷冻细胞之活化原则为快速解冻,以避免冰晶重新结晶而对细胞造成伤害,导致细胞之死亡。2、细胞活化后,约需数日,或继代一至二代,其细胞生长或特性表现才会恢复正常(例如产生单株抗体或是其它蛋白质)。3、材料37℃恒温水槽、新鲜培养基、无菌吸管/离心管/培养瓶、液氮或干冰容器4、步骤:(1)操作人员应戴防护面罩及手套,防止冷冻管可能爆裂之伤害。(2)自液氮或干冰容器中取出冷冻管,检查盖子是否旋紧,由于热胀冷缩过程,此时盖子易松掉。(3)将新鲜培养基置于37℃水槽中回温,回温后喷以70%酒精并擦拭之,移入无菌操作台内。(4)取出冷冻管,立即放入37℃水槽中快速解冻,轻摇冷冻管使其在1分钟内全部融化,以70%酒精擦拭保存管外部,移入无菌操作台内。(5)取出解冻之细胞悬浮液,缓缓加入有培养基之培养容器内(稀释比例为1:10~1:15),混合均匀,放入CO2培养箱培养。取0.1ml解冻细胞悬浮液作存活测试。(6)解冻后是否立即去除冷冻保护剂(例如DMSO或glycerol),依细胞种类而异,一般而言,大都不需要立即去除冷冻保护剂。惟若要立即去除,则将解冻之细胞悬浮液加入含有5-10ml培养基之离心管内,离心1000rpm,5分钟,移去上清液,加入新鲜培养基,混合均匀,放入CO2培养箱培养。(7)若不需立即去除冷冻保存剂,则在解冻培养后隔日更换培养基。三、细胞计数与存活测试1、原理:(1)计算细胞数目可用血球计数盘或是Coultercounter粒子计数器自动计数。(2)血球计数盘一般有二个chambers,每个chamber中细刻9个1mm2大正方形,其中4个角落之正方形再细刻16个小格,深度均为0.1mm。当chamber上方盖上盖玻片后,每个大正方形之体积为1mm2×0.1mm=1.0x10-4ml。使用时,计数每个大正方形内之细胞数目,乘以稀释倍数,再乘以104,即为每ml中之细胞数目。(3)存活测试之步骤为dyeexclusion,利用染料会渗入死细胞中而呈色,而活细胞因细胞膜完整,染料无法渗入而不会呈色。一般使用蓝色之trypan blue染料,如果细胞不易吸收trypan blue,则用红色之Erythrosin bluish。计算细胞活率:活细胞数/(活细胞数+死细胞数)×100%。计数应在台盼兰染色后数分钟内完成,随时间延长,部分活细胞也开始摄取染料;因为台盼兰对蛋白质有很强的亲和力,用不含血清的稀释液,可以使染色计数更为准确。2、材料:0.4%w/v trypan blue(GibcoBRL15250-061);Erythosin bluish stain;取0.1gram Erythrosin bluish(SigmaE-9259)及0.05gram preservative methyl paraben(SigmaH-3647)溶于100mlCa++/Mg++freesaline;血球计数盘及盖玻片(Hemocytometerandcoverslip);计数器(counter);低倍倒立显微镜;粒子计数器(Coultercounter,CoulterElectronics)。白细胞稀释液(4%乙酸溶液)。3、步骤:(1)取50μl细胞悬浮液与50μl trypan blue(orErythrosinbluish)等体积混合均匀于1.5ml小离心管中。(2)取少许混合液(约15μl)自血球计数盘chamber上方凹槽加入,盖上盖玻片,于100倍倒立显微镜下观察,活细胞不染色,死细胞则为蓝色(或红色-Erythrosin bluish)。(3)计数四个大方格之细胞总数,再除4,乘以稀释倍数(至少乘以2,因与trypanblue等体积混合),最后乘以104,即为每ml中细胞悬浮液之细胞数。若细胞位于线上,只计上线与右线之细胞(或计下线与左线之细胞)。注:4大格细胞总数×稀释倍数×104/4=细胞数/ml;每一大格的体积=0.1cm×0.1cm×0.01cm=10-4ml计数板计数时,最适浓度为5~10×105细胞/ml,此范围外计数误差偏大。高浓度细胞悬液,可取出部分作稀释或连续稀释后计数。5、范例:T75 monolayer culture制成10ml细胞悬浮液,取0.1ml溶液与0.1ml trypan blue混合均匀于试管中,取少许混合液加入血球计数盘,计数四大方格内之细胞数目。活细胞数/方格:55,62,49,59;死细胞数/方格:5,3,4,6;细胞总数=243平均细胞数/方格=60.75;稀释倍数=2;细胞数/ml:60.75×104×2(稀释倍数)=1.22×106;细胞数/flask(10ml):1.22×106×10ml=12.2×106存活率:225/243﹦92.6%







 我要推广仪器
我要推广仪器
 下载APP
下载APP