听说气相色谱可以测定样品中的微量水,很是好奇,不知道大家参考什么标准方法?测微量水,标准溶液大家如何配制呢?就用蒸馏水配吗?
成分分析中微量其他纤维的标准表示方法? 例: 1. 聚酯纤维:88% 2. 聚酯纤维:88% 3.聚酯纤维:88% 桑蚕丝:12% 桑蚕丝:12% 桑蚕丝:12% 含微量其他纤维 以上3种那个更标准一些?
谁有“压缩气体中微量油含量的测定标准” 测定的最低浓度为0.1mg/m3
求头发中钙铁锌等微量元素测定的国家标准方法或卫生标准方法
医院检测血液中微量元素有标准方法吗?是不是用的都是微波消解,血液中各种微量元素,如铜、铁、锌等为什么可以半天内就可以出结果?全自动生化分析仪真的如此神奇?
请问大家是如何称取微量的抗生素标准品的:用0.0000g的天枰称取5-10mg能准确吗?还有是能用药勺取药品吗?网上有人说使用药勺可能导致标准品污染
[font=新宋体][size=4][color=#DC143C]本人急求瓜果蔬菜中的微量元素的测定及其标准.....[/color][/size][/font]
微量金标准物质定值均匀性分析检验 利用王水溶样,活性炭吸附-灰化-王水溶解-硫脲提取-原子吸收光谱法测定微量金标准物质“GAu-9b”的30个子样的含量,每个子样重复测定3次。考察了天平称量精度、玻璃器皿刻度精度和活性炭吸附率造成的测量不确定度,并估算出合成不确定度范围为1.6%(A级品)~2.2%(A级品),论证了方法的可行性;对单个样品重复分析结果的可靠性及样品均匀性进行评估,计算出样品均匀性质量参数为0.77,样品测定结果的RSD均符合地质实验室质量管理规范要求,依此判定该样品的均匀性质量等级为合格。均匀性是标准物质必须具备的特性之一,均匀性检验得到的数据实际上包含了两种不确定度,一是样品不均匀引起的不确定度,另一是测量不确定度,所以在均匀性检验中应该使用高精度的方法,本文采用吸附效果好的活性炭吸附-灰化-王水溶解-石墨炉原子吸收光谱法测定微量金标准物质,以此方法为基础,结合石墨炉原子吸收对测定介质的要求,制定了微量金标准物质的分析步骤。通过查阅不确定度有关文献,计算了样品分析的合成不确定度,将合成的相对不确定度与重复分析相对偏差允许限进行比较,确定了拟定的分析方法的可靠性;均匀性检验方法很多,有方差法、极差法区间法和三分之一法等等 ,其中方差法和摄差法经多年的实践证明是可靠的,本文采用方差法(F)检验,得到样品均匀性良好;标准物质中各成分经均匀性检验合格后可进入定值,本文采用哥拉布斯 (Grubbs)法对离群值进行剔除,.最后得到标准物质的标准参考值和不确定度。1 实验部分1.1 仪器和主要试剂日立5000石墨炉原子吸收分光光度计,其测金条件略标准溶液:(ρ)=0.1μg/ml,工作液根据需要稀释,介质为Ψ=10%(体积分数,下同)的王水;盐酸、硝酸、硫脲试剂均为分析纯,水为蒸馏水;活性炭:粒径0.074mm,化学纯(北京光华木材厂[
微量元素液肥一个标准能不能做两种剂型,我想做粉剂和水剂两种
用原子分析光谱鉴别赭石所含各种微量元素,标准溶液的配置啊?翻阅了几个资料,都没有标准溶液的配置,那位帮帮忙啊?
各位大侠,谁那有《气体中微量水分的测定 电解法》这个标准,标准号是GB/T 5832.1-86.有的话能否提供一份,谢谢了!或直接发我信箱里,信箱地址是zhaoyu1016@126.com
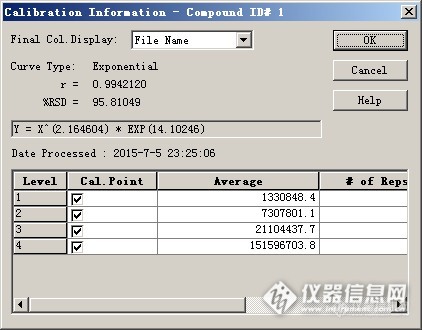
GC-FPD测定微量有机硫的标准曲线实验概述:GC-FPD测定微量有机硫的标准曲线建立方法。使用平方曲线和指数曲线的比对结果。我们知道,GC-FPD方法测定微量有机硫化合物的时候,FPD检测器的响应随浓度变化的趋势是非线性的。根据FPD的基本原理:含硫化合物分子进入检测器的富氢火焰中,形成激发态的S2分子,此激发态分子回到基态时会放出394nm的光,那么检测器的响应和进入检测器的硫化物的量的平方成正比。下面有一个分析实例:Shimadzu的GC-2014 -FPD,使用Rtx-WAX色谱柱,分析纯苯中微量的噻吩。标准品中噻吩浓度为0.89mg/l、1.77mg/l、3.54mg/l、8.9mg/l。进样后得到色谱图如下所示:http://ng1.17img.cn/bbsfiles/images/2017/10/2015070523363358_01_1604036_3.jpg使用GCsolution工作站,以峰高为定量依据,标准曲线类型选择“二次曲线”,权重因子设定为1/C2,绘制外标标准曲线,得到如下结果:相关系数和回归方程见下图。http://ng1.17img.cn/bbsfiles/images/2017/10/2015070523365896_01_1604036_3.jpg将标准样品数据回带入校准曲线,观察各个数据点的相对误差,均接近5%,可以认为该标准曲线拟合情况尚可。采用峰面积作为定量依据,其他条件相同的实验结果差距不大。如果仔细翻阅Shimadzu的FPD说明书,其建议绘制FPD-S标准曲线的时候,采用指数法。于是修改定量方法,重新处理数据,获得另外一条标准曲线。工作曲线的形式发生变化如下图。http://ng1.17img.cn/bbsfiles/images/2015/07/201507052337_553594_1604036_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507052337_553595_1604036_3.png然后将各个标准品数据点回带入标准曲线,发现各点的相对误差都发生变化,除去最高点误差稍大以外,其他数据点的相对误差均小于5%,级别1和级别2数据点的误差小于1%。标称浓度 0.89 1.77 3.54 8.86回代浓度 0.891 1.771 3.384 10.43(怀疑有问题)看来采用指数法的标准曲线,得到了更好的拟合效果(在低浓度区域是这样的,对于残留分析而言,这一点比较重要)。仔细观察一下,在本例试验中,FPD测定硫响应是浓度的2.16次方,而不是2次方。小结:其实常见检测器的响应都可以表示为Y=X的n次方的形式,只是常见的FID、TCD检测器的响应方程中n非常接近1。
气相色谱标准加入法测定环己酮中15种微量杂质 环己酮作为一种重要的化工原料和化工溶剂,是制造己内酰胺、己二酸和尼龙的重要中间体。目前环己酮的生产工艺主要有环己烷液相氧化,苯酚加氢、环己烯水合等多种方法,超过90%的环己酮是通过环己烷氧化的工艺生产的。工业用已内酰胺的产量逐年在增加,对环己酮的需求量也越来越大,随着已内酰胺高端市场的需求,对环己酮中杂质含量的控制要求严格。现有的环己酮产品检验方法主要依据工业用环己酮检验标准(GB/T10669-2001),采用填充柱分离,效果差。近年来,有关环己酮产品新的分析方法文献较少,对于环己酮中微量的环己烷、己醛、丁基环己烷、丁醇、4-庚酮、3-庚酮、2-庚酮、戊基环己烷、戊醇、环戊醇、2-甲基环己酮、3-甲基环己酮、4-甲基环己酮、环己醇等微量杂质达不到同时监控的效果,且分析误差较大。 本方法最终使用CP-Wax 52CB50m×0.32mm×1.2µm毛细管柱,程序升降温控制进行分离,采用外标法定量,同时测定环己酮中微量的环己烷、戊醛、己醛、丁基环己烷、丁醇、4-庚酮、3-庚酮、2-庚酮、戊基环己烷、戊醇、环戊醇、2-甲基环己酮、3-甲基环己酮、4-甲基环己酮、环己醇等15种杂质,分离度均大于1.5,且准确度高。1 实验部分1.1试剂与仪器 环己酮为南化公司生产,环己烷、戊醛、己醛、丁基环己烷、丁醇、4-庚酮、3-庚酮、2-庚酮、戊基环己烷、戊醇、环戊醇、2-甲基环己酮、3-甲基环己酮、4-甲基环己酮、环己醇为色谱纯。 Agilent7890气相色谱仪,FID检测器,Agilent Chemstaion色谱工作站,毛细管色谱柱 CP-Wax 52 CB 50m×0.32mm×1.2µm,1µl微量进样针。1.2 实验条件 汽化室温度250℃,检测器温度300℃,初始柱温115℃,程序升降温控制进行分离,柱流量0.8ml/min(恒流),分流比30:1,高纯He为载气,进样量为0.4µl。1.3[/f
请问检验铜合金里面的铜含量或者微量元素(比如铅等),用什么ICP仪器检测比较准?!!! 我们是一家铜合金产品制造公司,最近发现光谱分析里面的元素含量不是很准,想买一台ICP来测量元素含量,特别是微量元素等, 不知道买哪款仪器比较好? 请高人指点!!!
新手上路,望大家喜欢。Agilent1100微量标准制备自动进样器参考手册。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=33532]Agilent1100微量标准制备自动进样器参考手册[/url]
微量加样器是微量酶免疫测定中加样时必用的设备,其使用和校准对酶免疫测定结果有直接的影响。 加样器容量性能的鉴定,可依据国际标准化组织(1SO) 文件ISO/DIS8655和国家技术监督局颁发的有关定量、可调移液器的中华人民共和国国家计量检定规程《定量、可调移液器试行检定规程》(JJG646—90)规定的重量测试方法。这是目前用于此类仪器有效的校准方法。实验室可根据上述文件建立本室加样器校准的标准操作程序(SOP),下面是一个有关加样器校准的具体实例。 例:加样器校准标准操作程序1.适用加样器范围 各种品牌、型号的固定、可调和多通道加样器。2.校准方法3.校准环境和用具要求(1)室温:20—25℃,测定中波动范围不大于0.5℃。(2)电子天平:放置于无尘和震动影响的台面上,房间尽可能有空调。称量时,为保证天平内的湿度(相对湿度60%一90%),天平内应放置一装有10ml 蒸馏水的小烧杯。(3)小烧杯:5—10ml体积。(4)测定液体:温度为20~25~C的去气双蒸水。4.选定校准体积(1)拟校准体积;(2)加样器标定体积的中间体积;(3)最小可调体积(不小于拟校准体积的10%)5.校准步骤(1)将加样器调至拟校准体积,选择合适的吸头;如为固定体积加样器,则只有一种校准;(2)调节好天平;(3)来回吸吹蒸馏水3次,以使吸头湿润,用纱布拭干吸头;(4)垂直握住加样器,将吸头浸入液面2—3mm处,缓慢(1—3s)一致地吸取蒸馏水;(5)将吸头离开液面,靠在管壁,去掉吸头外部的液体;(6)将加样器以30角放人称量烧杯中,缓慢一致地将加样器压至第一档,等待压至第二档,使吸头里的液体完全排出;(7)记录称量值;(8)擦干吸头外面;(9)按上述步骤称量10次;(10)取10次测定值的均值作为最后加样器吸取的蒸馏水重量,按附表所列蒸馏水重量与体积换算因子计算体积。然后,按校准结果调节加样器。
刚看一个帖子说玻璃对微量元素有吸附,为什么我们买的标准溶液是玻璃瓶装的?
实验室搞认可,要查ICP-MS检测水中微量金属元素(Ba、Li、Ti、Cr等)的执行标准,实在找不到了,求高人指点下,谢谢!!
微量进样器(10ul-1000ul)拿去校准 看到校准证书上的依据是 JJG646-2006[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]检定规程。找到这个规程看下没发现微量进样器,这样用这个校准规程可以吗?
现使用1ml微量注射器检测空气中氧含量和氢含量,昨天原注射器破损了,请问使用新注射器该怎么校准呀!
各路英雄你们好,我需要确定样品中一种微量物质。我没有标准品,希望通过比对别人的质谱数据来确定。我想问,是不是质谱图各种离子碎片的丰度都要跟别人报道的一样才能确定呢??希望告知具体的确定方法,谢谢。

在紫外—可见分光光度计的使用中,标准比色池的样品容量是3毫升左右;当样品量很少且液面低于光轴时,单色器发出的用于测量光束(即光斑)照射不到样品溶液的侧面上,等于无法测量,尤其是萃取量很少的生化样品的分析。这点恐怕许多人都深有体会。见图-1所示:http://ng1.17img.cn/bbsfiles/images/2012/06/201206022301_370041_1602290_3.jpg图-1 用标准比色池测微量样品的示意要想解决这个矛盾,一般的情况下,是将平时经常使用的标准比色池改为微量比色池,如图-2所示:http://ng1.17img.cn/bbsfiles/images/2012/06/201206022302_370042_1602290_3.jpg图-2 改用微量比色池测试样品但是如果操作者手头一时没有微量比色池又怎么办呢?其实我们稍微动动手,问题则会迎刃而解。方法是:将一条宽度约为10毫米的白纸条插入到样品池的支架中;然后将波长调到530nm,此时会在白纸条上看到一个长方形绿色的光斑(注意:仪器周边环境要保持暗一些,否则不易看到绿光斑),并用笔在光斑处坐上记号,如图-3所示:http://ng1.17img.cn/bbsfiles/images/2012/06/201206022303_370043_1602290_3.jpg图-3 用笔在白纸条上的光斑处做上记号找一块黑色的海绵(泡沫塑料也可),根据光斑(即光轴坐标)与池架底部的垂直高度和池架内部的长宽,将海绵裁剪成为一个合适尺寸的块状物。需要注意的是:海绵的高度要略小于光斑高度,具体小多少,要依据样品量而定;这样做的目的,是给比色池的安放留有余地。见图-4所示:http://ng1.17img.cn/bbsfiles/images/2012/06/201206022305_370044_1602290_3.jpg图-4 裁剪海绵体将裁剪好的海绵安放到池架内,如图-5所示:http://ng1.17img.cn/bbsfiles/images/2012/06/201206031109_370064_1602290_3.jpg图-5 放置海绵体最后将标准比色池放置在池架内的海绵上,注入少量的溶液,以液面超过光轴坐标的垂直高度即可;见图-6所示:http://ng1.17img.cn/bbsfiles/images/2012/06/201206031113_370065_1602290_3.jpg图-6 安放标准比色池这种举措不但节省了溶液量和购置价格相对昂贵的微量池的费用,还有一个微量比色池不能比拟的优点,那就是:无需顾忌光斑与微量比色池透光槽的对位不良的问题。当然,这种举措也有它的局限性,就是样品量不能过少。为此、要根据所测试样品的平均量来剪裁海绵块的高度;海绵块的高度过高了,比色池同步也上抬了,光斑容易照到比色池的底部,造成测试误差;海绵块高度过低了,比色池随之下降,池子里注入的液体样品液随之增加了,这样则无谓地浪费了样品,没有体现出微量分析的初衷;因此做好二者高度的兼顾稍显麻烦些,需要有一定的耐心。谨记!
[font=Arial, 微软雅黑, 宋体][color=#505050]目前微量氧的分析一般采用氧分仪和[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分析,不论采用哪种分析方法,微量氧的分析采样是一个关键技术,因为空气中含有大量的氧气,如果取样过程出现任何小的纰漏都会造成数据的严重失真。取样阀一定要采用直通不带压力表的针形微调阀,这种阀体积小,置换起来比较方便,管线也一定要采用不锈钢硬链接的管线,任何软管都不能采用。[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分析微量氧的方法主要有TCD、ECD、DID以及氧化锆和质谱等方法,目前[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分析微量氧采用的色谱柱基本上都是填充柱,氧和氩气很难分开,这样就给TCD、DID等检测器分析微量氧带来了很大麻烦,因为我们国内生产的高纯氮气都含有2~上百ppm的氩气,国内生产标准气体的厂家基本上都没考虑高纯氮中氩的引入,这样很多客户在使用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]来分析微量氧时的数据完全失真,常常抱怨标准气体不准,因为高纯氮气中的微量氩很难除掉,用氦气或其他气体做平衡气,来解决这个问题,在必须采用氮气做平衡时,会给出氩气的含量作为分析微量氧的参考数据。[/color][/font]
检定测微量具标准器组建标技术报告[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=24427]检定测微量具标准器组建标技术报告[/url][color=red]【由于该附件或图片违规,已被版主删除】[/color]
在做HJ 734-2014这个标准时,要求用内标法定量,还需要加替代物,在进行吸附管加标时,我能用同一个微量进样针进行加标操作吗?
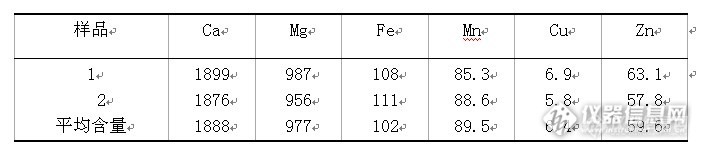
蕨菜中微量元素的测定【生活中的仪器分析】食品安全——“菜”米油盐酱醋茶大检测【摘 要】本文用湿法消解ICP-AES法测定了蕨菜中六种微量元素的含量。结果表明:蕨菜中Ca、Mg、Fe、Mn、Cu、Zn的含分别为:1899μg/g、987μg/g、108μg/g、85.3μg/g、6.9μg/g、63.1μg/g。【关键词】蕨菜 微量元素 ICP-AES蕨菜,为凤尾科植物,多年生草本植物,分布很广,除沙漠外,从高山至海关、从寒带到热带都有生长,常成片野生于荒坡、半山坡或林缘灌丛草地上。蕨根淀粉中含有丰富的蛋白质、维生素及人体所需的镁、锌、锗等微量元素。目前,蕨菜中有效成分在食品营养方面的应用已经成为关注焦点,大力研发蕨菜深加工产品将大有可为。国内在蕨菜毒理学方面的研究大多关注其功效成分的保健功能,国外有报道称,长期低剂量食用蕨菜可能引发膀胱肿瘤和间隙性牛血尿症,因此为了深入开发蕨菜等野生植物资源,有必要在急性毒性,亚急性毒性和慢性毒性方面对蕨菜等资源做更全面更深入的研究,为社会提供健康放心的食品。1、实验部分1.1、主要仪器和试剂仪器:ICP-AES、电热干燥箱、电子分析天平等试剂:硝酸、高氯酸标准储备液: 1.0mg/mlCa、Mg、Fe、Mn、Cu、Zn标准溶液 (国家标准物质研究中心) 1.2、ICP-AES工作条件http://ng1.17img.cn/bbsfiles/images/2013/12/201312231646_484160_2352694_3.jpg1.3、样品处理1.3.1样品的预处理将样品用自来水洗净,再用去蒸馏水漂洗,阴干,置于电热恒温干燥箱中,于50℃烘至恒重。随机取干燥蕨菜适量,用研钵研磨样品过40目筛,备用。1.3.2样品的消解准确称取样品0.5g(精确至0.0001g)两份,于100ml锥形瓶中,加入10ml浓硝酸、5ml高氯酸,放置过夜,盖上表面皿,在电热板上低温加热硝化处理,待溶液转为无色透明,冷却,转入50mL容量瓶中,用蒸馏水稀释至刻度,摇匀。2、结果与讨论2.1、分析元素波长经测定及最优化条件选择,确定出各元素的分析线波长见下表。http://ng1.17img.cn/bbsfiles/images/2013/12/201312231647_484161_2352694_3.jpg2.2、分析元素标准曲线的绘制http://ng1.17img.cn/bbsfiles/images/2013/12/201312231648_484162_2352694_3.jpg2.3测定结果经测定,蕨菜中六种元素的含量见下表http://ng1.17img.cn/bbsfiles/images/2013/12/201312231652_484163_2352694_3.jpg3.结论3.1、采用ICP-AES法测定蕨菜中的微量元素,表明除含有大量的Ca、Mg等常量元素外,还含有丰富的Fe、Mn、Cu、Zn等微量元素,其中Fe的含量最高。3.2、蕨菜中微量元素的协同和拮抗作用,使其成为不可多得的药食两用的植物。
[font=serif][color=#252525]测定油品中的水分可提供准确的计量油品的数量,即检尺后减去水量,就可得知整个容器中油的实际上数量。测出油品中的水分,可根据其含量的多少,确定脱水的方法,以防止造成以下危害:如石油产品中的水分蒸发时要吸收热量,会使发热量降低;轻质石油中的水分会使燃烧过程恶化,并能将溶解的盐带入气缸内,生成积炭,增加气缸的磨损;在低温情况下,燃料中的水会结冰,堵塞燃料导管和滤清器,阻碍发电机燃料系统的燃料供给;石油产品中有水会加速油品的氧化生胶;润滑油中有水时不但会引起发动机零件的腐蚀,而且水和高于100℃的金属零件接触时会变成水蒸气,破坏润滑油膜。轻质油品密度小,黏度小,油水容易分离。而重质油品则相反,不易分离。进入常减压蒸馏装置的原油要求含水量不大于0.2%~0.5%;成品油的规格标准要求汽油、煤油不含水,轻柴油水分含量不大于痕迹;重柴油水分含量不大于0.5%~1.5%;各种润滑油、燃料油都有相应的控制指标。[/color][/font][font=serif][color=#252525]所以测量润滑油或燃料油中的微量水分,该需要特定的仪器来进行,这是我们可以用常用的北京得利特的微量水分测定仪,他们家还有便携式的微量水分测定仪。外带也是十分方便。适用于很多标准且能广泛适用于石油、化工、电力、商检、科研、环保等领域。[/color][/font]

纺织品成分分析中含量1%以下与微量纤维怎么界定与判定[font='times new roman'] 纺织品纤维成分分析是[/font][font='times new roman']根据纺织纤维[/font][font='times new roman']的外观[/font][font='times new roman']纵[/font][font='times new roman']截面和横截面的形态特征和内在的不同性质,采用物理方法或者化学方法,辨别和区分各种纤维。通过各种实验来鉴别各种纺织纤维[/font][font='times new roman']。[/font][font='times new roman'],不仅用于单一纤维的定性,还用于鉴别及定量多种纤维混纺的纺织品[/font][font='times new roman']的纤维组成。[/font][font='times new roman'] 在常见的纺织纤维中,大多数都有了比较成熟的定量方法,比如[/font][font='times new roman']GB/T2910-2009[/font][font='times new roman']系列的检测方法,常规的纤维定量基本都能用到,其纤维定量的方法也比较成熟了,基本上按照纤维定性的结果选择适合的[/font][font='times new roman']检测方法进行检测即可。[/font][font='times new roman'] 纤维成分分析一般是取两个平行样,两个测试[/font][font='times new roman']样一起[/font][font='times new roman']进行前处理,需要褪色处理的要进行褪色处理,然后进行恒重,选择合适的分析方法溶解,干燥平衡,最后进行计算,两个平衡样的结果偏差不超过[/font][font='times new roman']1%[/font][font='times new roman'],即求两个试样的平均值为测试的最终结果。上报结果,成分分析完成。[/font] 但是最近遇见几个纤维计算后其中一种纤维含量再0.7-0.9%之间,均小于1%,这个值是按照标准溶解方法化学定量出来的,按照标准方法GBT 29862-2013纺织品 纤维含量的标识,进行出报告的话那么我这个样品成分定量结果是:50%聚酯纤维,49.3%棉,0.7%粘纤,按照标准方法检测和标示,我这个都没有任何问题,肯定也不算错,也是没有问题。 当时考虑到人员误差,试剂误差等等原因,最终把同一块样品送到省纤维检测院和市级纺织服装检测中心,省纤维检测院出的报告为50.5%聚酯纤维,49.5%棉(含微量其他纤维);市级纺织服装检测中心出的结果为:50%聚酯纤维,50%棉. 为了搞清楚他们的测试原理和方法是否和我们一至,经过多方努力终于联系到具体做这个适试样的两个工程师,省纤维检测院的工程师经确认我就是这个样品的送样人时,告诉我,如果按照溶剂法,几乎就没有微量纤维,两个试样溶解误差都不止0.5%,所以溶解微量纤维超过0.5%很正常的,哪怕这个纤维没有粘纤,你按标准进行溶解也会有百分之零点几的数据出来,所以一个试样溶解不超过1%的数据结果都是不可信的结果,一般都是出微量纤维,这个不是标准,是经验。 市级纺织服装检测中心的工程师告诉我,他们在显微镜下一个工程师能看到有1根粘纤,另一个工程师在显微镜下没有看到有粘纤,这个一般直接可以判定是微量,而且是不均匀的,按照[img=,636,103]https://ng1.17img.cn/bbsfiles/images/2021/08/202108031137589618_4128_2154459_3.png!w636x103.jpg[/img][font='times new roman'] 直接[/font][font='times new roman']出的结果为:[/font][font='times new roman']50%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']50%[/font][font='times new roman']棉[/font][font='times new roman'].[/font][font='times new roman'],这个是没有任何问题的。[/font][font='times new roman'] 通过这个样品,我们实验室内部也专门制定了一个作业程序,并进行了一个培训[/font][font='times new roman'],对成分分析做了以下几点[/font][font='times new roman']分析[/font][font='times new roman']。[/font][font='times new roman']1. [/font][font='times new roman']这个样品成分定量结果是:[/font][font='times new roman']50%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']49.3%[/font][font='times new roman']棉,[/font][font='times new roman']0.7%[/font][font='times new roman']粘[/font][font='times new roman']纤[/font][font='times new roman'];[/font][font='times new roman']50.5%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']49.5%[/font][font='times new roman']棉(含微量其他纤维)[/font][font='times new roman'];[/font][font='times new roman']50%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']50%[/font][font='times new roman']棉[/font][font='times new roman']三个结果都是正确的,[/font][font='times new roman']都不算错,但是结合实际情况,认为这个[/font][font='times new roman']50.5%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']49.5%[/font][font='times new roman']棉(含微量其他纤维)[/font][font='times new roman']是最合理的结果。[/font][font='times new roman']2. [/font][font='times new roman']成分分析定性要区多个试样,因为可能存在不均匀性,特别是纤维含量比较少的情况下[/font]3. GBT 29862-2013纺织品 纤维含量的标识,要多理解其中的说明,只要是符合其中的要求,就是可以的。
微量进样器的使用及保养: 微量进样器可供科研、化工、炼油、医院等单位作分析使用。特别适宜作[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]、[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]液体进行分析,是一种必不可少的精密器械。其总容量误差±5%。气密性能承受0.2mpa。为了达到延长进样器的使用寿命,进样器分为无存液与有存液两种,现将使用中应注意事项作以下说明:一、0.5μL-5μL无存液微量进样器 1.切忌重碱性溶液洗涤,以免玻璃受腐蚀失重和不锈钢零件受腐蚀损坏而引起漏水漏气。 2.本进样器是无存液之进样器。芯子(不锈钢丝)直接通到针尖端,故而不会出现寄存溶液,但使用后应立即清洁处理,避免芯子受污而卡死。 3.溶量指示芯子拉动不得超过AG图案,若不慎拉过图案和全部拉出外套而塞不进,可重新装配,但装配时必须先旋出针尖螺母,再耐心地把芯子(不锈钢丝)先从硅橡胶垫圈中心穿过,再对准针尖孔,慢慢旋上针尖螺母,再推进芯子,不准直接串入,以免不锈钢丝折弯。 4.本进样器的气密性依靠针尖部位的硅橡胶垫圈密封。故使用时期较长后,垫圈易损或老化会影响气密性,必须进行调换,否则,将会影响容量精度和重复性。 5.本进样器的针尖不得在400℃以上的高温下工作,更不宜用火直接烧,以免针尖退火而失去穿戳能力。
测定水中微量汞,做的标准曲线是0.1~1.0,最低点荧光值只有30不到,最高260左右,饮用水中汞的检测值要求达到0.05ppb,测定时好像很不稳定,差一点点荧光值,浓度就超标了,而且每次测定的值都不同.各位高手有没有什么好的方法提高标准的荧光值.我的仪器是吉天930.







 我要推广仪器
我要推广仪器
 下载APP
下载APP