高效液相色谱中,利用标准加入法测定内源性物质,方法学验证时,最低检测限如何确定?另外,回收率如何计算?
想请教各位前辈,刚开始做方法学就 碰到了难题,我是要测一些内源性物质,老板的意思是用同位素标记生物基质标准品配置工作曲线,那么这样的话,基质效应就会特别明显,因为标记的和未标记的是同时出峰,想请教一下各位前辈有没有什么解决办法,不胜感激!
各位前辈大家好: 初来乍到请多多关照!我最近想做监测鱼体内血清中类固醇激素的实验,实验室的仪器和设备都是很齐全的,我打算用HPLC-Q/TOF去做,先定性后定量。现在有一个问题难倒我了,就是标准品因为我做内源性的激素物质的监测,雄激素、雌激素和孕激素的标准品大多是哺乳动物和人的,我有3点疑惑,请懂的人帮忙指点一下:1、有卖的雌激素、雄激素和孕激素是从鱼体内提取的吗?谁知道请帮我提供一个公司的名称被 2、如果标准品都是从人或者哺乳动物体内提取的,我在用飞行时间质谱做鱼体内监测的时候能够扫描出来吗,比如说标准品是雌二醇是从人体内提取后制成标准品的,那么我监测鱼体内的内源性雌二醇能扫描出来吗,是同一种物质吗,分子结构是否相同呢?我看一些标准中检测过外源性性激素,他们所用的标准品是怎么合成的呢,我用这种标准品能够检测我鱼体内的呢?http://simg.instrument.com.cn/bbs/images/brow/em34.gif(说的有点麻烦,不知道大家看懂没) 3、用过ABSciex家的5600+的仪器的高手们,请问在没有标准品的情况下是否无法进行监测,不用标准品有没有什么软件或者技术能分析出来物质,没有标准品无法进行定量吧!问题很棘手也很多!请大家给予帮助,初来乍到积分很少,努力攒积分吧!谢谢了!
用液质测生物样品中内源性物质,要测多个组织?方法学验证时每个组织都要做一次精密度吗?把高中低浓度对照品加入相应组织?---您好,已转到 LCMS版(如不合适请版主帮忙转移)。下次发帖记得到对应的技术版面,新手版面的浏览量小 ,可能耽误您的问题。
生物样品中内源性物质——定量下限怎么算
[size=4] 在一定程度上,内源性的干扰一直困惑这很食品检测,在食品检测的残留级检测中更为严重,然而最为突出的就是内源性激素的干扰,很多人挠破额头皮也无法解决,回过头来发现自己的努力不过是在向着错误的方向进行,我建立不少激素方面的前处理方法,深受其苦; 原始的定义上讲来,内源性是内部本身就存在的东西,并不是外部干涉和添加,无法避免的; 然而在科学发达的今天,这个定义在部修改,人们可以通过对动物的阉割,使动物体内的雄性激素或雌性激素含量与生物界的本身产生极大的差异,从而获取利益,当然也有人给动物使用激素,例如给奶牛使用激素,使其不间断的产奶;这样的例子原来越多 听闻国家某部门宣称“圣元”奶粉中的内源性激素问题,我想问问,这些内源性真的是“内源性”吗?还是有人在“误解”这个词?大家说呢![/size]
今年鸡蛋例行监测中加了激素类参数的检测,结果发现每个鸡蛋样品中都检测出了黄体酮(孕酮),含量10-80ug/kg不等(查了一些相关文献,鸡蛋本身就含有黄体酮,属于内源性物质),而GB 31650中规定黄体酮为禁用药物,那么对于这类样品中本身就存在的物质如何判定是否是外源添加还是内源存在,这个临界值如何确定是很值得研究的,欢迎大家讨论。
[size=4][color=#DC143C][font=黑体]同位素比质谱方法检测内源性类固醇雄烯二酮[/font][/color][/size]=========================================在所使用的禁用物质中,类固醇激素是较为普遍使用的一类药物。人体自身能合成与分泌的类固醇激素称为内源性类固醇激素,如攀酮。由于在检测方法上有一定难度,一些运动员选择使用内源性类固醇制剂以逃避兴奋剂检测。目前,兴奋剂检测实验室应用同位素比质谱分析方法检测内源性类固醇来源。13C和12C是碳元素在自然界中的天然同位素。有机化合物的来源不同,其同位素比(如13C与12C的比值)也不同。人体自身分泌的类固醇与相同化学结构的类固醇制剂的同位素比不同。应用同位素比质谱分析技术可以测定化合物13C与12C的比值,同位素比用δ(‰)值表示。根据仪器的分析流程和组成部分,本文方法称为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比质谱([url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS)方法。该方法在兴奋剂检测中的应用时间较短,文献方法较为繁琐,本文建立了快速灵敏的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析方法。-------------------------------------------------试验材料与方法1. 试剂和对照品试剂:β-葡萄糖醛酸酶,Sigma;其余均为国产分析纯试剂。对照品:睾酮(缩写T)、雄酮(缩写An)、本胆烷醇酮(缩写Etio)、5α-雄烷-3α,17β-二醇(缩写5α-diol)、5β-雄烷-3α,17β-二醇(缩写5β-diol)、孕二醇(缩写PD)购自Sigma公司。2. 样品两名健康志愿者,一名男性40岁,尿样为sample 1;一名女性38岁,尿样为sample 2,均没有服用任何药物。收集其晨尿为阴性对照尿。阳性尿样为世界反兴奋剂机构水平考试所用尿样来自兴奋剂检测中心。3. 仪器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比值质谱仪(HP6890/DELTA PLUS,Finnigan);高效液相色谱仪(Waters2796,检测器:Waters2996 PAD,自动收集器:Waters Fraction Collector Ⅲ);Anilent 5973i[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用仪。4. 方法4.I [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS操作条件色谱柱:HP5毛细管色谱柱,25m×0.2mm i.d.×0.3μm;柱流速:1mL/min(室温);升温程序:60 ℃(2min)一50 ℃/min→255℃一2.5℃/min→280℃(6.5min);进样口温度:260℃;燃烧炉温度:960℃;质谱离子源:EI;参考气:CO2,1.8V。4.2 高效液相色谱仪操作条件色谱柱:ZQRBAX SB-C18(4.6mm×250mm,5μm);流动相:水-乙睛,梯度洗脱(0-18min:乙睛从30%→100%);流速1mL/min;柱温:室温。4.3 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD操作条件色谱柱:HP-1毛细管色谱柱,25 m×0.2mm i.d.×0.11μm;柱流速:1 mL/min (室温);升温程序:150 ℃(1min一5℃/min→200℃一30℃/min→310℃(10min)。4.4 样品预处理取尿样2mL,加人1Ml0.2mol pH=7.0的磷酸盐缓冲液和10μLβ-葡萄糖醛酸酶(5000 IU)混匀,在55℃恒温水浴中培养3h,取出后加pH=8.8的碳酸盐固体缓冲剂约100mg 和5mL叔丁基甲醚,振荡萃取,离心后,取出上层有机溶液,在加热的情况下,用氮气吹干,加人50μL甲醇溶解残渣,备用。将上述甲醇溶液置HPLC仪上,依前述色谱条件分离,分段收集流出液,确定收集时间程序。分别将流出组分用氮气吹干,加人50μL环己烷,备用。4.5 对照品溶液的制备分别配制对照品睾酮、雄酮、本胆烷醇酮、5α-雄烷-3α,17β-二醇、5β-雄烷-3α,17β-二醇,孕二醇的甲醇溶液,浓度为1mg/mL。4.6 样品测定4.6.1 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD 分析依上述[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD仪器操作条件,取样品溶液及对照品溶液进行全扫描,扫描范围m/z 20~450,选择待测物的特征离子,获得SIM图。经对样品中与对照品有相同保留时间的峰进行质谱分析,及与标准品质谱图的对比,确定待测样品的组成。4.6.2 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析依上述CC/C/IRMS仪器操作条件,对处理后的样品进行[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析,测得内源性类固醇激素的δ值。
想问一下用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]定量生物样本内源性的物质,怎么选择空白基质?基质加标做标曲
想请问大家关于如何确定生物组织体的内源性荧光组分,由于生物组织体有许多的能发光的内源性荧光物质,但是要如何一一区分开来呢,并且,我们对与一个单一组分的荧光物质的荧光光谱的带宽也是比较长宽的,如何说就是一个荧光物质在发光呢,请教大家了,欢迎e-mail to :lls009663@163.com.等待大家的答案!!!
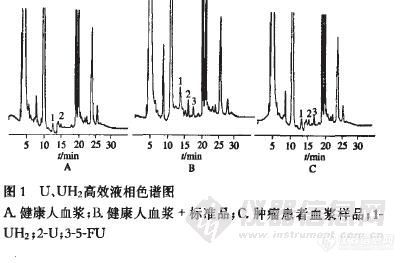
作者:肖力 任斌 陈小陆 李瑞明 刘怡 容颖慈 蓝缨(作者单位:中山大学附属第一医院药学部,广东,广州,510080 )摘要:目的:建立准确测定内源性尿嘧啶和二氢尿嘧啶血药浓度的高效液相色谱法.方法:以氟尿嘧啶(5-FU)为内标,醋酸乙酯-异丙醇混合液(85:15)为提取溶剂;色谱柱Diamonsil C18柱(250 mm×4.6 mm,5 靘);流动相A-0.01 mol·L-1磷酸二氢钾缓冲液(pH 5.5),B-乙腈,梯度洗脱;流速为0.8 mL·min-1;柱温为4 ℃;检测波长为204 nm(0~14.5 min),254 nm(14.5~35 min).结果:尿嘧啶和二氢尿嘧啶线性范围为8~500 靏稬-1,线性回归方程分别为C(UH2)=61.760 8Y+0.506 5,r=0.999 8;C(U)=95.201 1Y-3.064 0,r=0.999 3,(n=7).最低检测质量浓度均为5 靏稬-1.尿嘧啶方法回收率为99.3%~107.0%,二氢尿嘧啶方法回收率为95.0%~98.3%.尿嘧啶日内RSD小于6.5%,日阍RSD小于11.7%,二氢尿嘧啶目内RSD小于9.2%,日间RSD小于12.4%.结论:本方法可用于内源性尿嘧啶和二氢尿嘧啶血药浓度的常规监测.谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208142006_383845_1609970_3.jpg
除了要监控农药等有害物质的残留量,一些影响烟草品质的内源性组分如植物多酚、芳香胺等同时也要检测。植物多酚是烟草中的一类重要物质,其含量对烟草品质有重要影响,因而研究烟草及其制品中的植物多酚具有重要意义。通常的方法是用热甲醇水溶液对烟草中的多酚进行萃取,然后通过C18 SPE柱除去萃取物中弱极性脂类干扰物。①烟草样品中植物多酚的固相萃取方法烟样萃取:将烟样粉碎后过0.18 mm筛。取0.20 g,加入45 mL 80%(体积分数)甲醇水溶液,加热回流30 min。冷却后定容至50 mL。固相萃取净化SPE柱:Sep-Pak C18,Waters公司。柱预处理:30 mL甲醇,30 mL水,10 mL/min。样品过柱:5 mL上述甲水萃取液过柱,10 mL/min,弃去最初3 mL,收集后面2 mL。馏分过滤:0.45μm针头过滤器过滤,HPLC分析。②烟草中苯酚、儿茶酚固相萃取方法烟草中挥发酚在烟草燃烧后会产生不好的气味。苯酚和儿茶酚对呼吸系统有腐蚀和助癌作用。因此,近年人们开始关注并监控烟草中挥发酚的含量。以下是固相萃取方法:烟样萃取:20.00 g烟样,加入150 mL 2.5mol/L硫酸,加热回流l h,自动水气蒸馏仪以50%蒸汽量蒸馏15 min,收集其问的约230 mL馏分,加入1.7g磷酸二氢钾,调节pH至弱酸性,定容至250 mL。固相萃取净化SPE柱:Sep—Pak C18,30 mg,Waters公司。柱预处理:15 mL甲醇,30 mL 0.05 mol/L磷酸二氢钾缓冲溶液。样品过柱:50 mL上述样品过柱,10 mL/min。柱干燥:离心除水。目标物洗脱:2 mL 60%(体积分数)甲醇溶液。HPLC分析:Nova-pak C18色谱柱,流动相A-甲醇,B-0.05mol/L磷酸二氢钾缓冲溶液。梯度洗脱,0min 80%B,10 min 20%B,15 min 20%B,20 min80%&流速0.5 mL/min。检测器:DAD,进样量10mL。应用该方法对烟样进行萃取分析,回收率:苯酚91.8%~104.5%,儿茶酚89.6%~103.6%。来源:中国标准物质网

平时使用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]主要是用来测定各生物样本中各内源性代谢产物,经验不多,想跟大家分享一下尿样检测方法的建立过程遇到的一个很久才得以解决的问题。1、仪器Agilent 6890N gas chromatograph and Waters Micromass [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]T mass spectrometerDB-5 MS capillary column [img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911062032_182393_1056682_3.jpg[/img]2、样本预处理使用MSTFA对样本中各物质进行三甲基硅烷化衍生化处理,以提高样本中各物质的挥发性。3、遇到的问题在条件摸索过程中突然出现不出峰的现象。在尿样中含有多种内源性代谢产物,利用这台[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]T仪器,正常情况下至少可以检测到100个以上的谱峰,但在一次条件优化过程中突然无规律的出现出峰数量明显减少的现象,见图1。[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911062041_182397_1056682_3.jpg[/img]图1 出峰数量明显减少时得到的总离子流色谱图。分析后,总结造成这种情况可能的原因如下: 样品自身原因 进样问题 仪器自身原因 尿样中某些物质对仪器造成影响针对上述四种假设,一次排查。1)对同一份样本连续多次进样,见图2。[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911062041_182398_1056682_3.jpg[/img]图2 同一份样本连续进样四次,所得到的总离子流色谱图。这样可知,出现这种现象并非由样本预处理不好,未衍生化完全造成的。2)进样的考察更换进样人,反复尝试,依然存在此问题。3)仪器自身原因的排查 清洗衬管 更换隔垫 烘烤毛细管柱 清洗离子源完成上述处理后,重新进样考察,但仍然出现与之前类似的问题。其实做到这里是很郁闷的,整个排查过程总共用了很多天,但始终没有方向,感觉每一条路都被堵死,越来越混乱,也考虑了是否是样本中某种物质会干扰仪器,但由于这并不是最初的条件优化,之前很多次并没有出现问题。之后很偶然的想到了,是否是进样针出现了问题,因此特地买了支安捷伦进样针,没想到问题就这样解决了。其实,长时间使用的进样针会出现密闭不好的问题,这样吸取样本的量就受到很大的影响,最终导致了我上面所提到的问题,不知道各位的进样针多长时间更换一个,我是遇到了这个事才真正意识到这个问题。另外[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]进样针有两种,一种是锥形针尖的进样器,一种是锐尖针尖的进样器,但推荐使用锥形针尖的进样器,因为锐尖针头会扎碎进样口隔垫而导致泄漏,同时还因为其从隔垫抽出时被隔垫擦拭,从而引起色谱图上大的溶剂峰拖尾。如图3。[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911062042_182399_1056682_3.jpg[/img]图3 进样器针尖4、心得其实这个例子没有特别复杂的技术问题,讲给大家听只是想说其实在平时做实验遇到问题的时候,总是先想到比较复杂的、影响比较大的原因,而往往忽略了最简单的可能性。
各位好,我在用GC-MS(7890-5973c)检测茶叶鲜叶内源性挥发物质分析的时候,检测到了六氯乙烷,相对含量大概0.01%。因为样品不是很干,所以用冷冻干燥机干燥了大约4天,再用同样的提取及分析方法做,六氯乙烷的含量大概0.3%,而且还检测到了三氯乙烷和五氯乙烷。不知道这些有机氯化物是不是农药残留呢?另外,我的样品冷冻干燥后,用同样的方法检测,发现水杨酸甲酯及没食子酸变没了,而且邻苯三酚的含量却大幅提高了,不知道是什么原因,希望各位能多多给予知道,谢谢!
各位大侠,小女子最近用电化学检测器来测定人血浆中的儿茶酚胺,期见碰到了几个难题。(1)儿茶酚胺很不稳定,可以采取什麽措施来保证他的稳定性?(2)儿茶酚胺是内源性物质,如何得到不含儿茶酚胺的空白血浆用于标准曲线的制备?希望各位大侠提供些建议,不胜感激。
如果想用气质检测饮料里的挥发性物质,提取方法已经建立了,为了发外文文章,最好选国际上都知道的,应该选什么饮料呢,哪些饮料里有挥发性成分,大家给点建议吧
如果想用气质检测饮料里的挥发性物质,提取方法已经建立了,为了发外文文章,最好选国际上都知道的,应该选什么饮料呢,哪些饮料里有挥发性成分,大家给点建议吧
[size=4][font=黑体]苯甲酸属于自然界就存在的内源性物质,目前抽检到的含量水平对人体而言是安全的。当然,从预防原则出发,苯甲酸对特殊敏感人群的影响或许值得更多研究[/font][/size]广州市食品工业卫生检测所一项评估奶制品中苯甲酸含量的研究,经记者于2月17日首先报道之后,被网络以及平面媒体广泛转载,并引起了很大关注。 在国际期刊《食品管理》(Food Control)上,广州市食品工业卫生检测所戚平等技术人员报告了142份奶制品的分析结果,其中109份含有苯甲酸,其含量从0.51毫克/千克到110毫克/千克不等。 不过,一些媒体和公众对此研究结果产生误解,有人甚至认为,奶制品中根本就不应该出现苯甲酸这种物质。 对此,广州市食品工业卫生检测所技术人员希望重申,他们在奶制品中检测出的苯甲酸,属于内源性物质。也就是说,即使没有任何人为添加,也会或多或少地存在。 实际上,人们在空气、地表水以及土壤中,都发现过这种物质。在很多食物中,也都发现苯甲酸不同程度地存在。之前,世界卫生组织的报告显示,在中国,人们摄入苯甲酸和苯甲酸盐最主要的途径实际上是酱油等调味料。
RT现在用LC-MS测两个内源性物质,一个浓度接近定量限,一个浓度很高,出来的峰与邻近峰挤到一起(MS还是能识别开的),想请问一下大家,这种情况怎么处理?含量高的那个,我降低检测灵敏度行不行?因为低含量的那个存在,进样量,或浓缩比都不好降低了,而高浓度的这个实在太难看了,挤到一起,担心论文审核的时候会出岔子。谢谢大家!
各位老师好,我想请教一个问题:如何用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测出茶叶是否添加香精??我觉得最难点在于添加的香精未知,另外茶叶本身内源性香气组分非常多,那么即使添加香精也不好判断,因为内源性香气和外源性香精在仪器上检测到的组分是一样的。所以使用仪器的话还有什么办法可以检测判断是否添加香精呢?我不需要知道具体组分名称,只要仪器能够判断是否添加香精就可以了
有关物质检查,包括对产品中残留合成原料、中间体、副产物及可能的降解产物的检查,是控制药品质量的重要指标,目的是检查药品中所含的上述杂质是否符合安全性的要求,同时也是药品稳定性评价中需重点考察的项目。 有关物质检查常用的方法之一是HPLC主成分自身对照法(紫外检测器),即将HPLC色谱图中杂质峰面积与主成分自身对照液峰面积进行比较,以确定杂质限度是否合格。采用此方法时确定的检测波长是否合理直接影响到方法的可行性,因此检测波长的选择是方法学研究的重要内容。 在审评中发现一些申报单位在采用HPLC主成分自身对照法检查有关物质时直接或间接地以主成分的最大吸收波长作为检测波长,由于有关物质检查的对象是杂质,若将主药的最大吸收波长确定为检测波长,则杂质在此波长下的吸收可能偏低,某些杂质甚至无吸收,这样会造成对杂质含量的低估甚至漏检,从而不能反映产品的真实质量,影响了对品种质量可控性及稳定性的评价。1.直接将主药的最大吸收波长选作检测波长。2.简单地套用含量测定的色谱条件。在HPLC法进行含量测定时,为提高方法的灵敏度,降低干扰,往往选用主成分的最大吸收波长作为检测波长。若套用含量测定的色谱条件,实际仍是以主药的最大吸收波长作为有关物质检测波长。 3.以样品进行破坏性试验(酸、碱、热、光照、氧化等)后的溶液做紫外扫描,将扫描图谱中最大吸收波长确定为有关物质的检测波长。因破坏性试验后溶液中存在尚未破坏的主药、降解产物、辅料等,此溶液的紫外吸收为各成分紫外吸收的加和,并不能反映降解产物的紫外吸收特性。由于未破坏主药所占比例较大,故破坏性试验后溶液的最大吸收波长一般仍为主药的最大吸收波长。 采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近),选择检测波长时需对产品中可能存在的杂质(合成原料、中间体、副产物以及降解产物)的紫外吸收特性进行研究。已知杂质的紫外吸收特性可采用对其流动相溶液直接进行扫描的方法考察,未知杂质(如未知降解产物等)可通过二极管阵列检测器考察其紫外吸收情况,根据各主要杂质及主成分的紫外吸收特性,选取响应值基本一致的波长作为有关物质的检测波长。若对不同杂质难于找到均适宜的检测波长,可考虑选择在不同波长下分别测定,也可考虑采用加校正因子的主成分自身对照法。只有经试验研究确认主成分的最大吸收波长符合有关物质检查对测定波长的要求时,为方便操作,可选作有关物质的检测波长,以与含量测定的色谱条件一致。 另外,HPLC主成分自身对照法检查有关物质比较适用于对微量杂质总量的控制,也可用于单个杂质的限度(一般不超过0.5%)控制。对于具有明确归属的已知杂质,建议采用杂质对照品法进行检查。对于有毒有害杂质,更应采用杂质对照品法单独测定,并制定严格的限度。 转自——中国植提论坛
目 录1. 验证目的2. 验证范围3. 验证依据4. 验证时间5. 验证内容5.1 仪器配置5.2 验证所使用的试剂及标准品5.3 验证可接受标准5.4 需验证的测试方法5.5 系统适应性实验5.6 方法专属性5.7 系统精密度5.8 最小检测限5.9 最小定量限5.10 验证范围5.11 方法精密度5.12 线性范围5.13 方法准确度5.14 方法中间精密度5.15 方法耐用性6. 结果分析、结论及评价7. 附件 1. 验证目的确保--------相关物质检测方法准确、重现并耐用,为建立检验操作程序提供依据。2. 验证范围--------相关物质检测方法方法学验证。3. 验证依据ICH指南12.8(Validation of Analytical procedure);USP25版(1225)(Validation of Compendial Methods)。4. 验证时间2003年 月 日起至2003年 月 日5. 验证内容5.1 仪器配置名称型号序列号供应商校验日期校验结论有效时间高效液相色谱进样器[font=楷体_

放射性物质检查系统是一种专门用于放射性恐怖活动和放射性物质非法转移的新型安检系统。该系统采用非常稳定和可靠的探测材料,对放射性物质探测灵敏度高,具有结构轻巧,便于安装、携带,检测信息能经过有线或无线进行网络传输,实现远程控制与监测,是保护社会和公众免受放射性危害的理想检查系统。特点:1.系统结构简单,安装方便2.灵敏度高,对放射源方位有较好辨别能力应用范围: *机场、海关口岸*车站、码头*体育和会议场馆等用于监测和检查行人、行李等是否携带放射性物质*CIAE1108A型:与危险品检查通道相配合用于行李、手提箱的检测;*CIAE1108B型:与金属探测安检门配合用于非金属包壳的放射性物品检测。
我单位要加强对废旧金属行业的监管,需购进放射性物质监测的仪器,请问对于我们这专对于废旧金属行业放射性监测,我们买哪台仪器最好呢.
有关物质检查方法的验证重点关注专属性、灵敏度和耐用性,以及杂质的相应因子。⑴ 专属性 专属性应确保杂质从主成分峰中分离出来或各杂质与主成分峰互相之间应分离,并且分离度应符合要求。 原料药可用合成反应(特别是后三步合成反应)的原料、中间体,粗品、重结晶母液等考察杂质与主成分的分离情况。仅采用破坏性试验考察,可能会漏掉合成原料、中间体、立体异构体等杂质,应当引起注意。同时配合不同柱效的色谱柱、不同波长(或用DAD全光谱检测)和不同流动相比例来考察。 破坏性试验适合于考察降解产物,更适合于制剂有关物质检查方法的验证。一般用酸、碱、光、热、氧化反应等加速破坏性来验证,应当注意破坏程度不宜太大,一般以使主成分降解5~10%为好,同时应该充分注意质量平衡原则,防止分解产物无法被检出的情况发生。用过期或变质的样品来验证,也是较好的选择。杂质峰响应值与主成分峰的不一致、杂质峰无法检出或被破坏等均能引起质量不平衡。 如果有液相色谱—串联质谱联用仪,可以对主成分峰进行质谱扫描,以验证主成分峰是否还包含杂质。DAD 的峰纯度指数一般不能作为主成分峰是否单纯的验证方法,因为杂质的量很小对峰的贡献很小,对峰纯度指数的影响也很小,即便纯度指数指示为纯峰并不能代表主成分峰不包含杂质。再则如果主成分峰与杂质峰完全重迭或杂质的光谱与主成分相似,报告的峰纯度指数仍然是很高的。⑵ 灵敏度 灵敏度是有关物质检查的重要指标。灵敏度应满足测定所关注杂质的要求,至少应达到所关注杂质限量的10%。由于不同的高效液相色谱仪的灵敏度不相同,建议在标准中设立或检验时配制灵敏度试验溶液,灵敏度试验溶液的浓度一般不大于供试品溶液的 0.05%,杂质量大的可以浓一些,杂质量小的应当稀一些。灵敏度溶液主峰的信噪比应当不小于10。⑶ 耐用性 耐用性一般可以考察三个以上品牌色谱柱、流动相比例变化±2%~5%、流动相 pH 值变化±0.2、柱温变化±5℃、检测波长变化±5nm。在上述条件下,均能较好地满足设定的系统适用性试验要求。 ⑷ 单个杂质的响应因子在具有杂质对照品时,考察杂质相对于主成分的相应因子以及色谱峰的相对保留时间,并观察其稳定性。 ⑸ 溶液稳定性试验除特殊情况外,应在室温下考察。主要应关注杂质峰个数、位置及其峰面积与杂质总峰面积是否发生变化。
准备用酶促荧光法测粪便样品中的D-乳酸含量, 原理:D-乳酸在D-乳酸脱氢酶(D-LDH)的催化下生成丙酮酸,同时把NAD+转变为NADH,NADH具有荧光特性,通过测定NADH 的浓度达到间接测样品中 D-乳酸盐的浓度。已知未去蛋白的样品易受到内源性的L-LDH和D-LDH的干扰,因为NAD+是这两个酶的共同底物。为了避免非特异的NAD+转化成NADH而导致D-乳酸盐浓度的高估,对粪便样品进行前处理非常必要。查文献说,用高氯酸脱蛋白,用量与样品体积比为1:1。存在疑问:用高氯酸脱蛋白用量应怎么确定?如果用量不足,不足以除去蛋白(内源性酶),如果用量过高,可能对其后加入的酶具有破坏作用,怎样才能确定一个合适的用量,即能保证除去样品中的内源性酶,又不至于影响后来加入用来反应的酶,请各位高手帮忙解决一下这个问题?谢谢!如果不用高氯酸脱蛋白,用其它的试剂也存在同样的问题。
各省、自治区、直辖市食品药品监督管理局(药品监督管理局),有关单位: 为进一步加强化妆品安全监管工作,规范化妆品中禁限用物质检测方法,现对有关单位研究起草的化妆品中挥发性有机溶剂等禁限用物质检测方法(征求意见稿)公开征求意见,请将修改意见于2011年1月4日前反馈我司。 联 系 人:林庆斌 联系电话:010-88330884 传 真:010-88373268 电子邮件:linqb@sfda.gov.cn 附件: 1、《化妆品中挥发性有机溶剂检测方法》(征求意见稿) 2、《化妆品中钕等15种稀土元素检测方法》(征求意见稿) 3、《化妆品中邻苯二甲酸酯检测方法》 (征求意见稿) 4、反馈意见表 国家食品药品监督管理局食品许可司 二〇一〇年十二月二十二日
物质检测方法
——胡丽涛、王治国目的 为凝血酶原时间(PT)和活化部分凝血酶时间(APTT)检测提供指南。方法 主要参考美国临床和实验室标准化研究院(CLSI)H47-A2文件相关内容,并结合具体的实际进行总结。结果 PT和APTT检测受到检验前、中和后各种变异的影响。通过检测技术的标准化提高检测的重复性,通过设定性能目标来确保检测的临床相关性。PT检测可用于检测肝素治疗,实验室应该建立肝素治疗范围。结论 对于疑似血液凝固障碍的患者评估,PT和APTT是非常重要的实验室筛查检测。在实际工作中要严格按照操作规程和厂家说明,保证结果的准确可靠,为临床提供诊疗依据。凝血酶原时间;部分活化凝血酶时间;国际标准化比值;校准 自从Quick在1953年首次描述了凝血酶原时间(PT),PT在可疑凝血障碍的患者评估方面成为了一项非常重要的实验室筛查检测,也是实验室最常见的凝血检测。尽管PT最先被描述成一种凝血酶原或凝血因子Ⅱ的特异性一步法检测,但是PT对凝血系统的共同途径和外源性途径所涉及到的任何凝血因子(凝血因子Ⅱ、Ⅴ、Ⅶ、Ⅹ和纤维蛋白原)及因子抑制物的定量或定性的异常都非常敏感。同时,PT也是严重肝脏疾病或慢性肝病的一项适当指标,还是用于监测维生素K治疗最常用的指标。 PT检测的主要试剂凝血活酶是一种磷脂或组织制剂,通常为人源性或动物源性或人和动物的混合材料制成的各种商业化的制剂。这些商业化的凝血活酶制剂在其减少凝血因子的反应性方面存在差异,影响了其使用,特别是在维生素K治疗的检测过程中。 APTT对内源性凝血途径和共同途径的定量和定性异常敏感。是除PT之外,常规实验室最常进行的凝血检测。APTT对内源性凝血途径凝血因子缺乏(凝血因子Ⅷ、Ⅸ、Ⅺ、Ⅻ、前激肽释放酶和高分子激肽原)尤为敏感。APTT检测通常用于监测低分子肝素抗凝治疗。APTT还用于检测其他类型的病理型血液凝固抑制物,最常见的是狼疮抗凝物质(LA)。APTT用于凝血因子替代治疗。APTT的检测试剂是促凝物、磷脂和接触激活剂组成的混合物。磷脂可能来源于人、动物或者植物。激活剂的种类也很多,如硅藻土、高岭土、微粉化的硅土和鞣花酸等。 一般,当凝血因子的活性低于参考区间的95%置信限水平时APTT会延长。但是,大量的研究表明不同的APTT试剂对于轻度和中度的凝血因子缺乏的反应程度存在一定的区别,特别是Ⅷ和Ⅸ因子缺乏。有相似的报道APTT对循环狼疮抗凝物质有不同的敏感性。同样,有文献报道商品化的APTT试剂对肝素的反应性不同,使得APTT值存在显著差异。
准备用酶促荧光法测粪便样品中的D-乳酸含量, 原理:D-乳酸在D-乳酸脱氢酶(D-LDH)的催化下生成丙酮酸,同时把NAD+转变为NADH,NADH具有荧光特性,通过测定NADH 的浓度达到间接测样品中 D-乳酸盐的浓度。已知未去蛋白的样品易受到内源性的L-LDH和D-LDH的干扰,因为NAD+是这两个酶的共同底物。为了避免非特异的NAD+转化成NADH而导致D-乳酸盐浓度的高估,对粪便样品进行前处理非常必要。查文献说,用高氯酸脱蛋白,用量与样品体积比为1:1。存在疑问:用高氯酸脱蛋白用量应怎么确定?如果用量不足,不足以除去蛋白(内源性酶),如果用量过高,可能对其后加入的酶具有破坏作用,怎样才能确定一个合适的用量,即能保证除去样品中的内源性酶,又不至于影响后来加入用来反应的酶,请各位高手帮忙解决一下这个问题?谢谢!如果不用高氯酸脱蛋白,用其它的试剂也存在同样的问题。







 我要推广仪器
我要推广仪器
 下载APP
下载APP