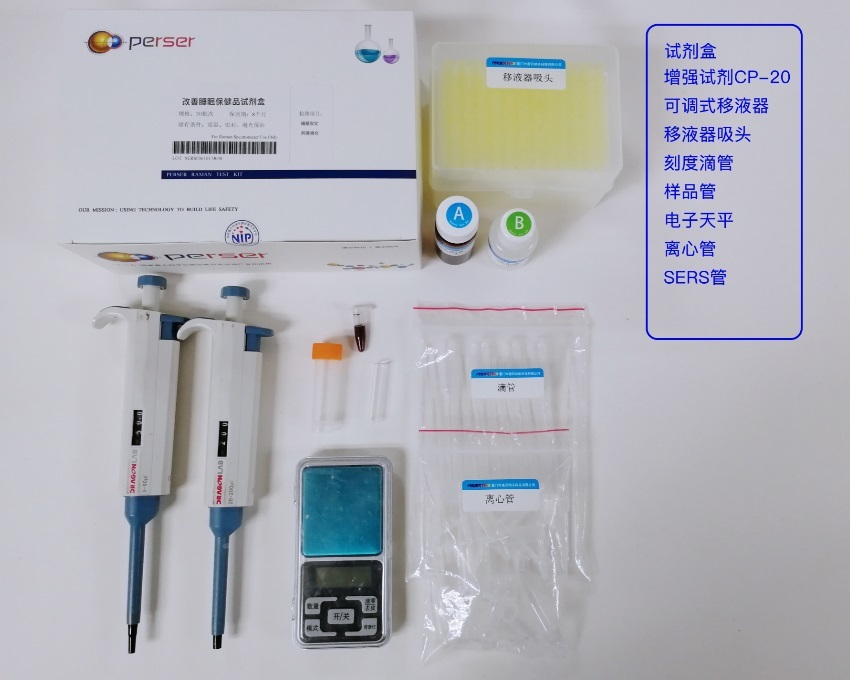
推荐厂家
暂无
暂无

 400-860-5168转3662
400-860-5168转3662
 留言咨询
留言咨询
 400-860-5168转3662
留言咨询
400-860-5168转3662
留言咨询
 留言咨询
留言咨询


进口药品注册标准号字母JX、JF有什么意义和区别吗?
[size=3][b]国家药品标准包括的类别探讨[/b][/size]国家药品标准包括:1)中国药典2000年版、2005年版;中国药典2002、2004年增补本; 中国药典2005年版勘误; 2006年、2009年增补本; 2)卫生部中药成方制剂一至二十册、二十一册(中药保密品种); 3)卫生部化学、生化、抗生素药品第一分册;4)卫生部药品标准(二部)一册至六册;6)卫生部药品标准藏药第一册、蒙药分册、维吾尔药分册; 7)卫生部新药转正标准1至75册; 8) 国家药品标准化学药品地标升国标一至十六册;国家药品标准化学药品地标升国标一至十六册勘误;9)国家中成药标准汇编内科心系分册、内科肝胆分册、内科脾胃分册、内科气血津液分册、内科肺系(一)、(二)分册、内科肾系分册、外科妇科分册、骨伤科分册、口腔肿瘤儿科分册、眼科耳鼻喉皮肤科分册、经络肢体脑系分册;10)国家药监局和国家药典委员会颁布的新药批件及修订批件;11) 进口药品复核标准汇编;12)进口药品单页标准不知除此之外还有什么标准吗,望补充
药品质量标准论坛网 http://standard.joinbbs.net/附件为注射用尼麦角林进口注册标准











 我要推广仪器
我要推广仪器
 下载APP
下载APP