求助:利用原子荧光光度计(AFS-921)检测样品中的总砷,采用的是标准曲线法,同一批次样品中,总有一两个样品的荧光值超出标准曲线。荧光值出现*号,如:*3620.120。出现这样的数据结果该如何处理?求大神指教。
样品中浓度超出线性范围时大家都如何处理?a)减少样品量重新处理?b)或者原先样品量处理的最后步骤稀释萃取液?c)还是一般都有保存的萃取液,稀释保存萃取液重新定量?
我ICP瓦里安710的,最近几天发现当样品中被测元素超出曲线范围就显示不出结果,我的情况不是如“2546x”这种情况,我知道这样显示是浓度太高的原因,问题是现在显示"点点点x"就是“……x”,没有数字,只有点,所以还请大虾们讨论一下,谢谢!
我记得有个标准里解释了浓度超出曲线最高点,为何要稀释至曲线中间点附近,但是标准名忘了,求告知
我们一般做生活污水厂、医疗废水,都采用样品蒸馏的方法,然后再进行纳氏试剂比色法进行比色!按照国标(HJ 535-2009 ),需要取250ml的样品,蒸馏至约200ml,再定容至250ml。1、可是,我们污水浓度太高,我就取了50ml 样品,再蒸馏至约200ml,再定容至250ml,不知道这样是否合理?还是说取50ml 样品,再蒸馏至约40ml,再定容至50ml合理呢?2、蒸馏后的样品浓度依然超出我的标准曲线的浓度范围,我是不是需要对蒸馏后的水样进行稀释呢?3、污水是否可以直接采用絮凝沉淀的方法,再稀释倍数后,进行测定的结果好呢?还是蒸馏后测定的结果好呢?哪个的准确度更好呢?请各位专业人士和同仁给予指导!谢谢啦!
请问你们ICPAES法测定17种金属元素时,返测QC溶液(我们一般是用中间浓度点的标准溶液来作QC溶液),所测得结果的偏差范围要控制在多小,如果QC溶液的结果超出了控制范围,你们是如何处理的??我们客户要定期检查我们测试的原始数据以及QC溶液的控制情况的。有时候QC的结果超出了控制范围,要重新做曲线,很费时间,样品数量又很多,测试报告出具时间也很赶,有时真的快把人急坏了,不知道你们有什么好的办法或者一些不合常规的捷径,能把QC的数据测得准确些,尽量在控制范围内。
我ICP瓦里安710的,最近几天发现当样品中被测元素超出曲线范围就显示不出结果,显示“……x” ,以前都不会,哪位大虾教教小弟是怎么回事呀~!
请教下,你们做有组织VOCs的工作曲线范围是多少?TENAX管采集的样品浓度超出曲线范围了,如何出数?
请问你们用GCMS法测定8P时,返测QC溶液(我们一般是用中间浓度点的标准溶液来作QC溶液),所测得结果的偏差范围要控制在多小,如果QC溶液的结果超出了控制范围,你们是如何处理的??我们客户要定期检查我们测试的原始数据以及QC溶液的控制情况的。有时候QC的结果超出了控制范围,要重新做曲线,很费时间,样品数量又很多,测试报告出具时间也很赶,有时真的快把人急坏了,不知道你们有什么好的办法或者一些不合常规的捷径,能把QC的数据测得准确些,尽量在控制范围内。
超出内控标准但符合国家批准的产品,如何处理?多数公司都会制定一个高于国家标准的内控标准,但当成品的检查结果,超出内控标准,但符合国家批准的标准时,该如何处理该批成品?需要走什么手续?
如果生产出的药品其中一项指标超出了企业内控,但是却满足国家标准,是否可以放行?如何进行?需要哪些手续?希望大家把各自执行的情况说一下,谢谢![em61]
超出原标准 适用范围是否属于对方法的偏离?
我用外标法作标准曲线,由于标准样品是液态,所以进样时注入1ul标准浓度的溶液。求出标准浓度和其相应面积的关系曲线。但是在实际分析样品时用顶空进样法,进样量大概1ml。这样在分析样品时如何使用标准曲线。内标法在该样品中不太合适。或者其他定量方法能更好地解决这个问题。希望您能不吝赐教。衷心感谢。
采用液相法检测浓度为mg/kg级的样品,单点校正时,标准品浓度与样品浓度相差不超过多少为宜,该规定出自哪里?
单点校正时,标准品与样品的浓度相差不能超过30%,这30%出自哪里?
作为实验室分析人员,除了掌握仪器分析技能外,还要了解标准的使用和执行相关知识。如,某个检测标准规定“本标准适用于苹果、柑橘、葡萄、甘蓝、芹菜、茄子中农药及相关化学品残留量的测定,其它蔬菜和水果可参照执行”。该标准的适用范围只规定了这些试材,如果需要检测韭菜,那么就超出了该标准的使用范围,需要对该标准进行方法确认。提示:是确认,而不是验证。
如何考虑测试样品基体的干扰?我在用ICP-OES测试铜合金ROHS项目时,铅的含量达到35000ppm左右,超出了我所建立的标准曲线浓度范围,如何考虑测试过程中是否有样品中基体的干扰?请各位老师帮忙指点一下,谢谢!
[font=仿宋_GB2312][size=21px][color=#6b6b6b]土壤中半挥发性有机污染物上机溶液的仪器测试结果不允许超出标准曲线上限,当仪器测试结果超出标准曲线上限时,可采取以下方式进行分析:([/color][/size][/font][font=仿宋_GB2312][size=21px][color=#6b6b6b]1)重新萃取样品后稀释萃取液;(2)将样品充分均质后,减少取样量。[/color][/size][/font]
样品浓度过高,吸光度虽然超出了标准曲线,但也可以代入标线上算出值来,为什么得稀释样品呢?
液相色谱进样量一般多少微升?我样品浓度不高,进样3-5微升,出峰总平头,超出检测器吸收极限。
超出预定范围使用的标准方法,在能力表述时,何种情况表述为标准方法,何种情况表述为自编方法(标准作业程序SOP)?
[align=center][b]校准曲线5个典型问题解答[/b][/align]1[align=center][color=#ff0000][b][size=14px]标准曲线查出样品是负值,如何报告结果呢?[/size][/b][/color][/align][font=宋体][size=15px][color=#00d100][b][/b][/color][/size][/font][font=宋体][size=15px][color=#000000]这种情况下我们一般选择四种方式报告结果:未检出,0.05的最高的那个浓度点为止。有也经验说当加入的高浓度点在低浓度点构建的直线方程推算的响应超出±10%范围。关于线性范围的最低点应该是0、检出限或是定量限的问题,个人趋向最低点为0,因为线性范围只是统计学上线性的表现,跟能否准确定量是两方面的问题。[/color][/size][/font][font=微软雅黑][size=12px][color=#000000]来源:实验室ISO17025[/color][/size][/font]
您的实验室在进行检测时,是否会遇到以下情况?平行双样超出标准允许偏差范围遇到这种情况一般如何处理?如果您采用的是进行第二次检测,那么第二次检测的平行双样再次超出标准允许偏差范围,你又是如何处理呢?(以上情况均出现在正常检测情况下,也就是说所有检测过程均按照作业指导书进行!故排除操作出现不符合检测工作的情况)
请问如果要建立多点一次的内标曲线,标准样品和内标物的浓度怎么配啊?昨天我赔了几个不同浓度的标样,但内标物的浓度基本没有改变,只是改变了标样的浓度,可最后为什么都不呈线性呢?点乱取八糟的,不明白是为什么?难道是改变标样浓度的同时也要改变内标物的浓度吗?请高手多多指点。谢谢了!多点一次就是配制不同的浓度,每个浓度进样一次,我想用不同浓度的标准品来建立标准曲线,以前用的是同一个浓度重复进样建立的内标曲线。我现在只是把待测的物质配成不同浓度,而内标物的浓度基本不变,可做出来的曲线怎么就不呈线性了呢?是不是内标物的浓度要跟着待测物仪器改变吗?但是那样的话又和同意浓度有什么区别呢?
配制了具有浓度梯度的一系列标准溶液,用HPLC测峰面积时,0号管(浓度为0ug/ml)有峰面积,我绘制标准曲线时,将所有标准溶液管的峰面积都扣除了0号管峰面积,然后绘制了标准曲线,请问求样品浓度时,样品的峰面积也需要扣除0号管峰面积吗?
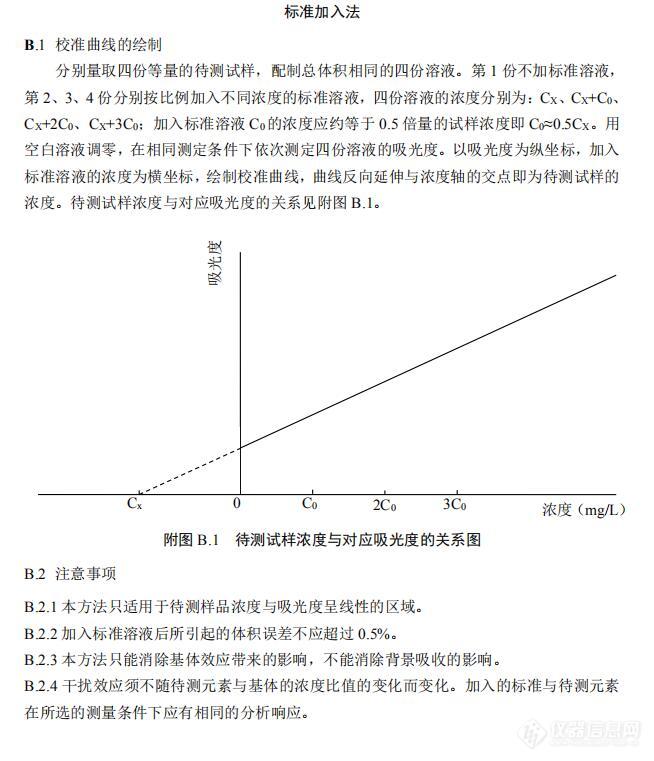
1、样品的吸光度值必须是在标准曲线的中部吗,假设标准曲线的浓度范围是0.1mg/L-2mg/L,当样品的吸光度值接近标曲的0.1mg/L,甚至略低时适用吗,未见权威资料对C[size=10px]0样品浓度提出要求[/size]?2、标准加入有下面两种提法,哪种更适用,火焰方式下,样品(3-5g无机样品溶解于50mL溶液)的吸光度值可能只有0.01-0.02abs,无法达到下述的A1在0.1-0.2abs,即样品A0的吸光度需要达到0.05-0.1abs,如果非要达到这个吸光度,那只能提高5倍左右样品浓度,这么高的浓度,燃烧头一下就堵死了。摘自:HJ 749-2015 固体废物 总铬的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[img=,690,709]https://ng1.17img.cn/bbsfiles/images/2020/06/202006191659437494_7814_1638724_3.jpg!w690x709.jpg[/img]摘自:分析化学手册(第三版)3A 原子光谱分析[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/06/202006191657599964_1720_1638724_3.jpg!w690x517.jpg[/img]
情况:前一天进样时,很正常 今天,(同样的试剂,同一瓶贮备液,同一种方法),用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]进样,前一天的浓度都没出锋,进了一针比平时高100倍浓度的标准品,出现拖尾,找不到原因,请版友们指点
各位大虾,帮忙分析一下。我用的是PE2100(ICP-OES),在做标准样品(QC)时所有的元素浓度都偏高20%左右,这是什么原因呢?而且用5%HNO3冲洗几分钟后,又正常了。标准曲线是刚做的,线性非常好,而且做的那个标准样品也是标准曲线中的一个点。雾化效率,泵速,提升量的影响应该和标准溶液的浓度没关系吧?是不是进入等离子体的样品越多,其强度也会越强啊?其发光强度是与进入等离子体的样品的浓度还是质量成正比呀?
[align=center][b]校准曲线5个典型问题解答[/b][/align]1[align=center][color=#ff0000][b][size=14px]标准曲线查出样品是负值,如何报告结果呢?[/size][/b][/color][/align][font=宋体][size=15px][color=#00d100][b][/b][/color][/size][/font][font=宋体][size=15px][color=#000000]这种情况下我们一般选择四种方式报告结果:未检出,0.05的最高的那个浓度点为止。有也经验说当加入的高浓度点在低浓度点构建的直线方程推算的响应超出±10%范围。关于线性范围的最低点应该是0、检出限或是定量限的问题,个人趋向最低点为0,因为线性范围只是统计学上线性的表现,跟能否准确定量是两方面的问题。[/color][/size][/font][font=微软雅黑][size=12px][color=#000000]来源:实验室ISO17025[/color][/size][/font][size=18px][color=#00bbec]声明:[/color][/size][color=#888888]部分图文信息来源互联网,版权归属原作者。本公众号为学习交流平台,并不用于商业目的。如有侵权请及时告知,经核实后删除;[/color]
如题。用空白基质(如血浆、尿液等)稀释样品后测定,在标准曲线范围内,但是在建方法时稀释比怎么做,要怎么考查?







 我要推广仪器
我要推广仪器
 下载APP
下载APP