有用不同分流比来稀释配置标准曲线或做校正因子的吗?之前做标准曲线从来没想过用不同分流比来配标准曲线,不了解分流过程的准确度如何现在是做高纯混合气体分析,需要定期做相对质量校正因子,但自己配制标准气体有点麻烦,想直接用不同分流比(对应到不同样品的组分浓度)来算校正因子
请问实验室间比对时,不同实验室可以按照不同标准检测吗? 还是必须用同一标准? 因为存在检测项目相同,但是有不同的标准。
大家谈一谈实验室或检测中心内部不同分工人员的收入分配问题,或提些建议[em0808]
请问实验室间比对时,不同实验室可以按照不同标准检测吗? 还是必须用同一标准? 因为存在检测项目相同,但是有不同的标准。
[table=100%][tr][td]用带有紫外检测器的高效液相色谱检测物质,同分异构体能有不同的峰吗?能互相分离吗?[/td][/tr][/table]
通过两种不同分析原理的监测分析方法测定地表水中总镉含量比对实验,实验表明利用HSJ-Cd型总镉在线监测仪测定地表水中镉含量具有在线监测快速准确、干扰少、分析成本低、信息反馈迅速等优点;在线自动监测系统监测结果的精密性、准确性较好,相对标准偏差(RSD)小于8.6%,相对误差小于5.7%,回归方程相关系数大于0.999,能够满足环境监测质量的要求,对及时迅速、科学准确掌握地表水环境质量状况,实现环境自动监控具有重要意义。
我们实验室使用23200.113检测果蔬样品已经两年了,但是一直有几个疑问没能解决,与其他人咨询过,但是各家说法有些不一致,所以想向各位大神咨询一下答案。像氟虫腈,甲拌磷,倍硫磷这类化合物,在限值标准2763中写的是:以氟虫腈/甲拌磷/倍硫磷表示,但是结果要包含2个以上同分异构体的含量,这样就涉及到不同化合物结果加和,关于加和方法我有以下3个疑问(以氟虫腈为例):1:氟虫腈检出限为0.005mg/kg,如果各同分异构体单独含量都不达检出限,但是加和后能达到检出限以上,这种情况是加和报检出 还是 不加和报未检出?2:若某个样品,氟虫腈的有部分同分异构体含量不达检出限,但是加上它们后,检出值超过了这个样品的国标限值,这个样品应该算超标还是算未超标?3:同分异构体检出后,以检出浓度计算得出了各个异构体的含量,做加和计算时,需不需要考虑它们之间的质量换算系数?比如氟虫腈亚砜换算成氟虫腈,氟虫腈相对分子质量437.15,氟虫腈亚砜相对分子质量421.15。以上问题期待收到各位专家的答复,感谢!?
近期,做样过程中发现,同一浓度样品所有条件相同情况下,FID检测器响应值增加了近两倍,且基线高于20pA。解决过程如下:1.对仪器进行常规维护:清洁进样针、更换隔垫、清洗衬管和分流平板、清洗FID、老化色谱柱。常规维护完成后,重新开机进样测试,基线下降恢复正常,样品响应值增加的问题依然存在。2.改变分析方法参数,确定故障原因:将原来的分流比进样更改成不分流或者不同分流比进样,发现更改分流比对响应值竟然没有影响,初步确定故障原因大概是分流出口管线堵塞。于是将进样口和分流捕集阱之间的管线拆卸下来,发现的确是管线堵塞,更换管线后问题解决。[img]https://ng1.17img.cn/bbsfiles/images/2021/03/202103071911395503_6326_3015727_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/03/202103071911395952_2016_3015727_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/03/202103071911396979_7324_3015727_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/03/202103071911406783_3371_3015727_3.png[/img]
不同进样方式(分流与无分流)对饱和烃的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分布特征及相关地球化学指标的影响 原文:邹宇峥 蔡元明 岩石抽提物和原油中饱和烃的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]特征是石油地质和地球化学工作者最为基础的研究依据。但是,无论是提供这些研究依据的实验人员还是应用这些研究依据的地球化学工作者或石油地质工作人员普遍地会忽视[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]不同进样方式(分流与无分流,以及分流时的不同分流比)对饱和烃[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分布特征及相关地化指标的影响。本文通过对饱和烃系列标样(nC9、nC11、nC13、nC15、nC17、nC19、nC21、nC23、nC25、nC28、nC30)和典型的实际饱和烃样品在不同进样方式下的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分布特征及相关地化指标的研究,得到以下结论:① 不同进样方式对饱和烃[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分布特征的影响是显著的。这主要表现在分流方式下饱和烃中低碳数正构烷烃轻组分不同程度的丧失,即所谓的“歧视现象”,使色谱分布形态完全改变,甚至包括主峰碳的改变;② 正构烷烃标样的结果:如果假定无分流方式下,所有样品全部进入到毛细色谱柱中。并以它为标准,以碳数作为横坐标,分流方式下各种不同分流比实验所得到的不同碳数饱和烃的含量都与无分流方式实验的对应碳数的含量相比所得到的比值作为纵坐标(图1),可以看到:分流比越大,所得到的轻组分相对含量越低,从而导致重组分含量的相对增加;如果均以最大碳数为标准,对数据进一步归一化处理,其差别将会更显著地表现出来(图2):分流比越大的曲线越远离无分流方式的直线;③ 实际样品的结果:与正构烷烃标样的结果几乎一样,只是低碳数区域有反常现象(图3、图4)。可能的原因:在实际样品中,低碳数正构烷烃的含量很低,瞬间汽化所造成的“歧视现象”不仅与宽沸程样品中各组分的沸点有关,还与各组分的实际含量有关。即,高含量轻组分将优先被“歧视” ,低含量轻组分的“歧视现象”不如高含量轻组分的“歧视现象”明显;④ 相关地化指标的影响:Pr/Ph值在各种进样方式下的变化不大,相对标准偏差为4.54%。解释:对于沸点相近的组分,“歧视作用”的影响程度也相近。另外,无分流方式下的(ΣnC21-) / (ΣnC21+) 值为4.824, 而分流比为50:1时的(ΣnC21-) / (ΣnC21+) 值为2.287,相差了两倍多,这一影响是决对不能忽视的;⑤ 本文同时也做了特大分流比(100:1、200:1)的实验,在如此大的分流下,其实验数据与无分流方式相比,无上述规律可寻,有很大的随机性。解释:在进样体积相同的情况下,采用特大分流比时,进入到色谱柱中的样品只有原来的一百分之一和二百分之一,瞬间汽化所造成的样品不均一性的可能性极大,进入到色谱柱中的样品已经完全和实际样品的真实组成大相径廷;因此,数据提供者应尽量避免使用特大分流比做色谱实验,最好使用无分流进样方式。研究者在利用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]数据时,首先必须注意到色谱所采用的分流方式。不同进样方式下,色谱数据的某些地化指标,如(ΣnC21-) / (Σn C21+),是没有可比性的,绝对不可以混用。而对于另外一些地化指标,由于与指标相关的各组分的沸点相近,由不同进样方式导致的“歧视作用”程度接近,进样方式对这些指标的影响是可以忽略的。同时建议新的饱和烃[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]国标方法应该对分流比进行统一规定,最好统一规定使用无分流进样方式,以便使[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]数据具有广泛的可比性。图1 系列正构烷烃标样在各种大小不同分流比时的各组分含量与 无分流进样方式对应碳数含量的相对值对比图(SP代表无分流)图2 系列正构烷烃标样在各种大小不同分流比时各组分含量与 无分流进样方式对应碳数含量的归一化后的相对值对比图(SP代表无分流) 图3 某实际饱和烃样品中的正构烷烃在各种大小不同分流比时各组分含量与 无分流进样方式对应碳数含量的相对值对比图(SP代表无分流) 图4 某实际饱和烃样品中的正构烷烃在各种大小不同分流比时各组分含量与 无分流进样方式对应碳数含量的归一化后的相对值对比图(SP代表无分流) 摘录:四海男儿(发表时间:2002-2-5 23:28:13)
检验检测机构的质量控制方法主要包括以下几种?: ?定期使用有证标准物质进行监控?:使用有证标准物质或内部标准样品作为监控样品,定期或不定期将监控样品与样品检测以相同的流程和方法同时进行,验证检测结果的准确性?。 ?参加实验室间的比对或能力验证计划?:通过与其他实验室进行比对或参与能力验证计划,评估本实验室的检测能力?。 ?利用相同或不同方法进行重复检测或校准?:使用不同分析方法或同一型号的不同仪器对同一样品进行对比检测,确保检测结果的可靠性?。 ?对存留物品进行再检测或再校准?:对保留样品进行再次检测或校准,以监控检测过程的稳定性?。 ?人员比对?:由实验室内部的检测人员在相同条件下对同一样品进行检测,比较检测结果的符合程度,评估检测人员的操作能力和稳定性?。 ?方法比对?:不同分析方法之间的比对试验,比较不同方法的检测结果,验证方法的可靠性?。 ?设备比对?:使用不同设备对同一样品进行检测,比较结果的符合程度,确保设备的准确性?。 ?加标测定回收率?:在样品中加入已知浓度的标准物质,测定回收率,评估检测过程的准确性?。 ?环境质量控制?:确保实验室的环境条件满足检测工作的需要,监测和控制环境因素对检测结果的影响?。 ?[back=linear-gradient(to right, rgba(111, 75, 250, 0.12), rgba(111, 75, 250, 0.12)) center bottom / 100%12px no-repeat]设备和仪器的质量控制?:定期对设备进行检定(校准),并在两次检定期间进行期间核查,确保设备的准确性和稳定性?。
向各位老师请教,机动车检测比对试验如何实施?如何对检测结果进行分析?为什么会提这个问题,主要原因有以下几个方面,1、没有测量的参考值,在不同的线间或站间不知道哪个结果更接近真实值。2、比对试验时不可能进行多次测量,一般一条线也就3次左右,有些项目多次测量,怕对对车辆产生损伤,如动力性、环保检测等。3、比对试验时影响因素太多,检测结果差异会比较大。如前照灯检测时,每次停车位置对偏移量的影响;制动检测时,脚踩踏板力的大小,对制动力大小也有直接的影响。这些影响因素在比对时往往很难克服,那该如何做比较好呢?请各位指教!
有点搞不懂, GCMS设定不同的分流比会哪些影响?比如对出峰强度会否有影响.什么情况下使用不分流,什么情况下使用20:1分流比,检测的结果会有影响吗?谢谢
我们化验室里一直都是用的快速法测定COD的,就是取完样品后机器上165度消解10min,等冷却后加水混匀,再等冷却后在分光光度计上610nm处测出结果,最近上面领导要求我们用国标法和快速法做比对,结果两者做出来的数值有相差。 我们水样的cod浓度一般是20-80,做的时候我自配了个浓度50的标液带着一起做,结果快速法标液检测值49,高(进水)67,低(出水)34;而国标法标液检测值52,高(进水)53,低(出水)20,现在的问题是两种方法测定的标液值都在误差范围内,但是水样差别比较大,都是快速法的结果高于国标法(前面也有几次检测,都是快速法的结果高于国标法),国标法不是经常做,但完全按照步骤来的,之前每次有环保所的质控样来就会做,做出来也基本接近真值的,国标法用的是低浓度的,水样取样前都摇晃混匀的,进水ss做下来有四十几,求有经验的前辈帮忙分析一下,看看哪个环节出了问题,先谢谢了。
1 质控过程方法比对是不同分析方法之间的比对试验,指同一检测人员对同一样品采用不同的检测方法,检测同一项目,比较测定结果的符合程度,判定其可比性,以验证方法的可靠性。方法比对的考核对象为检测方法,主要目的是评价不同检测方法的检测结果是否存在显著性差异。比对时,通常以标准方法所得检测结果作为参考值,用其他检测方法的检测结果与之进行对比,方法之间的检测结果差异应该符合评价要求,否则,即证明非标方法是不适用的,或者需要进一步修改、优化。2 适用范围方法比对主要用于考察不同的检测方法之间存在的系统误差,监控检测结果的有效性,其次也用于对实验室涉及的非标方法的确认。整体的检测方法一般包括样品前处理方法和仪器方法,只要前处理方法不同,不管仪器方法是否相同,都归类为方法比对。但是,如果不同的检测方法中样品的前处理方法相同,仅是检测仪器设备不同,一般将其归类为仪器比对。
NMR对同分异构体可以定量检测吗?? 谢谢!怎样检测啊??
NY/T 1977-2010标准中氮含量检测根据氮种类不同分为几个检测方法在这几个方法检测过程中若用的试剂都是一样的话那这空白值是否都是一个值我目前在总氮和硝态氮、 铵态氮空白都是一样的今天测脲态氮空白值没有做平行样空白值偏低不知道是操作问题还是理论就偏低

上个月,给客户安装的圆二色检测器。此检测器可同时检测圆二色和紫外信号。现贴图如下,CH1的是圆二色信号,CH2的是紫外信号。http://ng1.17img.cn/bbsfiles/images/2011/08/201108081111_309115_2041919_3.jpg同时可以样品的光谱扫描。下图的两个光谱图,为上面同分异构体的光谱图。http://ng1.17img.cn/bbsfiles/images/2011/08/201108081118_309116_2041919_3.jpg
不同厂家的农残速测仪检测同一样品的结果误差是多少为合格?如何进行仪器之间的比对?在选好的空白样品加农药吗?加多少为合适?如何计算回收率呢?谁做过这样的试验写出来共享吧。
想检测同分异构体混合物的总含量怎么检测?比如松油醇同分异构体混合物。
大家好,我们参加了一个实验室间比对的实验,其中Na含量检测结果出了问题,但经过调查、复测都没法找到原因,想请大家一起讨论看看是哪里出了问题,具体情况是这样的:方法是[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]火焰法,样品是营养液(类似于乳制品一类的),样品预处理是用的干法消化;初次(参加对比的结果)60.40mg/100g;实验室间比对的平均值是44.75mg/100g;我们偏差很大,于是就调查和复测,测下来的结果是这样的第一次复测:47.31mg/100g;第二次复测54.74mg/100g;三次检测有3个不同的结果,第一次复测的结果还是接近的。不知道是哪里出了问题。我们觉得可能有这样几个原因造成的1) 是样品不均匀;2) 检测过程被污染了但找不到具体的问题出在哪里,请大家一起讨论分析一下。谢谢!
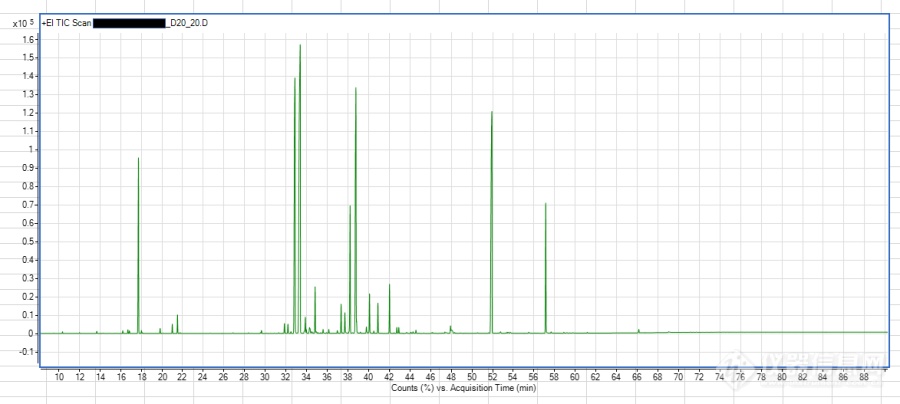
之前香精样品直接50:1 分流比进样,浓度太高, 尝试20被稀释, 100:1,50:1 20:1, 10:1, 5:1 分流比进样情况如下:[img=,690,305]https://ng1.17img.cn/bbsfiles/images/2022/03/202203281112042696_1848_5536964_3.png!w690x305.jpg[/img][img=,690,300]https://ng1.17img.cn/bbsfiles/images/2022/03/202203281112222451_1906_5536964_3.png!w690x300.jpg[/img][img=,690,309]https://ng1.17img.cn/bbsfiles/images/2022/03/202203281112431792_9123_5536964_3.png!w690x309.jpg[/img][img=,690,305]https://ng1.17img.cn/bbsfiles/images/2022/03/202203281112585403_3901_5536964_3.png!w690x305.jpg[/img][img=,690,305]https://ng1.17img.cn/bbsfiles/images/2022/03/202203281113140206_3458_5536964_3.png!w690x305.jpg[/img]
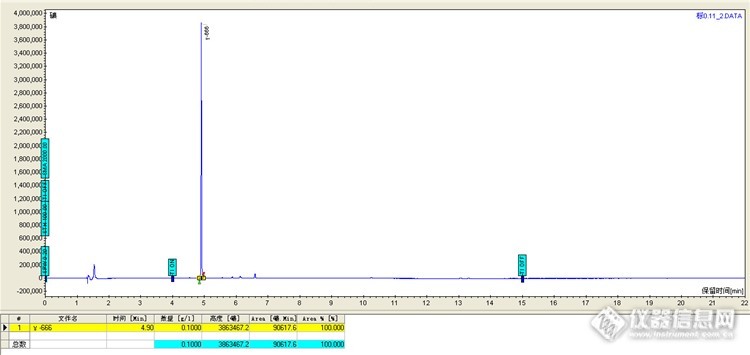
第36期草根比对-γ-666的检测 4月份我与同事都报名参加了第36期草根比对,γ-666的检测,写出来检测过程,分享一下。 1 实验部分 1.1 仪器与试剂 GC450气相色谱仪(天美公司),PFPD、ECD检测器,自动进样器 1.2实验方法 稀释盲样后直接进样。http://ng1.17img.cn/bbsfiles/images/2016/05/201605101549_592802_1645480_3.jpg两支安瓿瓶盲样,1.2mL。1.3 标准溶液的配置由农业部环境保护科研监测所提供,质量浓度均为100ug/mL,有机氯的标液用正已烷稀释至10.0ug/mL,继续稀释到0.1ug/mL,进样测定峰面积。http://ng1.17img.cn/bbsfiles/images/2016/05/201605101549_592803_1645480_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/05/201605101549_592804_1645480_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/05/201605101549_592805_1645480_3.jpg正已烷溶剂的标液γ-666,浓度为100ug/mL。1.4实验条件有机氯的检测条件色谱柱:VF-1ms(30m×0.32mm×0.25um);温度:80℃保持1min,以15℃速度上升到280℃,保持10.00min。进样体积:1uL;进样品:250℃,不分流进样;检测器300℃;进样口温度:250℃,氮气流速:30mL/min;柱流速:1mL/min。1.5实验过程1.5.1盲样吸取1.0mL到10mL容量瓶里,定容到刻度,混匀,吸取250uL到10mL容量瓶里,定容到刻度,混匀,进样测峰面积。http://ng1.17img.cn/bbsfiles/images/2016/05/201605101550_592806_1645480_3.jpg盲样进6针,RSD《3%,重现性很好,记录峰面积。http://ng1.17img.cn/bbsfiles/images/2016/05/201605101550_592807_1645480_3.jpgγ-666标液0.1ug/mL的色谱图。2 结果与讨论2.1计算含量 用盲样的峰面积/标液峰面积*标液浓度(0.1ug/mL),等于盲样的含量。2.2计算稀释倍数 吸取盲样1.0mL到10mL容量瓶里,稀释倍数为10,吸取250uL到10mL容量瓶里,稀释倍数为0.025,所以稀释倍数为0.0025。2.3配相近点的标液 从以上可知盲样的含量,为了更准确些,可配制与盲样相近的标液点进行比较,比如盲样的含量为0.40ug/mL,这时需要配制0.40ug/mL的标液进样确定峰面积,看两者的差异。3结论通过做此次草根比对,有几点可以总结一下:3.1盲样稀释用移液枪,吸了1.0mL,没有把盲样全部倒入容量瓶里稀释,因为不知盲样的1.2mL是否准确。3.2气相要准备好,用新色谱柱、新进样垫、新衬管,提前一夜开机,预热ECD,这样基线会很平稳。3.3此次盲样检测用的是单点校正,单点校正的方法简单,但如果浓度相差较大,会出现较大的误差,所以先用标液0.1ug/mL来计算盲样大概含量,再配制相近的标液点来计算,这样就提高了准确性。
分流进样是毛细管气相色谱的首选的进样方式,适用于大部分气体或液体样品的分析,尤其对未知样品或已知“较脏”的样品,使用分流进样,可保护毛细管柱,减小或避免色谱柱被污染的可能。该进样方式进样时,进入进样口的样品大部分被分流掉而没经过色谱柱,仅有少量的样品进入毛细管柱。分流比就是分流流量与柱流量的比值。例如,如果进入色谱柱的流量就是1 mL/min,分流流量为20 mL/min,那么分流比是20︰1。实际工作中,我们应该如何测定分流比和分流量?分流比对物质的分离测定又有何影响?
[color=#333333]1.分流管线的清洗:[/color][color=#333333] [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]用于有机物和高分子化合物的剖析时,许多有机物的凝固点较低,样品从气化室经过分流管线放空的进程中,部分有机物在分流管线凝固。[/color][color=#333333][url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]经过长期的运用后,分流管线的内径逐步变小,甚至彻底被阻塞。分流管线被阻塞后,仪器进样口显现压力反常,峰形变差,剖析成果反常。在检修进程中,无论事前能否判别分流管线有无阻塞现象,都需求对分流管线进行清洗。分流管线的清洗一般挑选丙酮、甲苯等有机溶剂,对阻塞严峻的分流管线有时用单纯清洗的办法很难清洗洁净,需求采纳一些其他辅佐的机械办法来完结。能够选取粗细适宜的钢丝对分流管线进行简单的疏通,然后再用丙酮、甲苯等有机溶剂进行清洗。因为事前不容易对分流部分的状况作出准确判别,对手动分流的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]来说,在检修进程中对分流管线进行清洗是十分必要的。[/color][color=#333333]关于EPC控制分流的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],因为长期运用,有可能使一些细微的进样垫屑进入EPC与气体管线接口处,随时可能对EPC部分构成阻塞或构成进样口压力改变。所以每次检修进程尽量对仪器EPC部分进行检查,并用甲苯、丙酮等有机溶剂进行清洗,然后烘干处理。[/color][color=#333333]因为进样等原因,进样口的外部随时可能会构成部分有机物凝结,可用脱脂棉蘸取丙酮、甲苯等有机物对进样口进行开始的擦洗,然后对擦不掉的有机物先用机械办法去除,留意在去除凝固有机物的进程中一定要当心操作,不要对仪器部件构成损害。将凝固的有机物去除后,然后用有机溶剂对仪器部件进行仔细擦洗。[/color][color=#333333][/color][color=#333333]2、TCD和F ID检测器的清洗[/color][color=#333333]TCD检测器在运用进程中可能会被柱流出的沉积物或样品中夹藏的其他物质所污染。TCD检测器一旦被污染,仪器的基线呈现颤动、噪声添加。有必要对检测器进行清洗。[/color][color=#333333]HP的TCD检测器能够选用热清洗的办法,具体办法如下: 关闭检测器,把柱子从检测器接头上拆下,把柱箱内检测器的接头用死堵堵死,将参考气的流量设置到20 ~ 30 ml/min, 设置检测器温度为400,热清洗4~8 h,降温后即可运用。[/color][color=#333333]国产或日产TCD检测器污染可用以下办法。仪器停机后,将TCD的气路进口拆下,用50 ml打针器依次将丙酮(或甲苯,可依据样品的化学性质选用不同的溶剂)无水乙醇、蒸馏水从进气口反复注入5~10次, 用吸尔球从进气口处缓慢吹气, 吹出杂质和剩余液体, 然后重新安装好进气接头, 开机后将柱温升到200 , 检测器温度升到250 , 通入比剖析操作气流大1~2倍的载气, 直到基线安稳为止。[/color][color=#333333]关于严峻污染, 可将出气口用死堵堵死, 从进气口注满丙酮(或甲苯,可依据样品的化学性质选用不同的溶剂) ,保持8 h左右,排出废液,然后按上述办法处理。[/color][color=#333333]FID检测器的清洗: F ID检测器在运用中安稳性好,对运用要求相对较低,运用遍及,但在长期运用进程中,容易呈现检测器喷嘴和搜集极积炭等问题,或有机物在喷嘴或搜集极处沉积等状况。对FID积炭或有机物沉积等问题,能够先对检测器喷嘴和搜集极用丙酮、甲苯、甲醇等有机溶剂进行清洗。当积炭较厚不能清洗洁净的时分,能够对检测器积炭较厚的部分用细砂纸当心打磨。留意在打磨进程中不要对检测器构成损害。开始打磨完结后,对污染部分进一步用软布进行擦洗,再用有机溶剂zui后进行清洗,一般即可消除。[/color][color=#333333][/color][color=#333333]3、打针器的清洗[/color][color=#333333]打针器运用前可先用丙酮清洗,以免玷污样品,但zui好还是用待打针样品对打针器自身做一二次清洗。清洗时只能吸入样品,排出样品时要在样品瓶之外。打针器在运用完毕后要当即清洗,以免被样品中的高沸点物质玷污。一般常用下述溶液依次清洗:5%氢氧化钠水溶液、蒸馏水、丙酮,zui后用真空泵抽干。[/color][color=#333333]用于有机物和高分子化合物定量剖析的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]一般选用分流进样,毛细管色谱柱。依据仪器的生产厂家和类型的不同,进样口的分流控制系统一般有EPC控制分流和手动控制分流两种状况。[/color][color=#333333]在检修时,对[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]进样口的玻璃衬管、分流平板,进样口的分流管线, EPC等部件别离进行清洗是十分必要的。[/color][color=#333333]玻璃衬管和分流平板的清洗: 从仪器中当心取出玻璃衬管,用镊子或其他小工具当心移去衬管内的玻璃毛和其它杂质,移取进程不要划伤衬管外表。[/color][color=#333333]假如条件答应,可将开始清理过的玻璃衬管在有机溶剂顶用超声波进行清洗,烘干后运用。也能够用丙酮、甲苯等有机溶剂直接清洗,清洗完结后经过干燥即可运用。[/color][color=#333333]分流平板zui为抱负的清洗办法是在溶剂中超声处理,烘干后运用。也能够挑选适宜的有机溶剂清洗:从进样口取出分流平板后,首先选用甲苯等惰性溶剂清洗,再用甲醇等醇类溶剂进行清洗,烘干后运用。[/color]
我们一直用PS聚苯乙烯做GPC的窄标,可是最近的实验让我很疑惑:我同时(所用仪器条件都一样)进两组不同分子量的PS(为了方便理解,将其分别命名为A组A1、A2、A3、A4,B组B1、B2、B3、B4,而且A组和B组对应的分子量都很接近,也都分布在我们样品结果范围周围)和一个样品X,做完后分别用这两组标样去处理这个样品,发现处理结果竟然相差五六千(如用A组标样校正得到样品X的MW是97000,而用B组标样得到是92000,重复实验都是这样)!很是疑惑!那我应该相信哪个结果呢?GPC是相对较正的结果,我觉得标样的这点差异不至于对结果有那么大的影响吧?而且阿我们每次购买标样都不可能是与上次分子量相同的,这样的数据不是没多大的可比性?请求各位帮忙解惑啊!在下先感激了![em0808]
我们一直用PS聚苯乙烯做GPC的窄标,可是最近的实验让我很疑惑:我同时(所用仪器条件都一样)进两组不同分子量的PS(为了方便理解,将其分别命名为A组A1、A2、A3、A4,B组B1、B2、B3、B4,而且A组和B组对应的分子量都很接近,也都分布在我们样品结果范围周围)和一个样品X,做完后分别用这两组标样去处理这个样品,发现处理结果竟然相差五六千(如用A组标样校正得到样品X的MW是97000,而用B组标样得到是92000,重复实验都是这样)!很是疑惑!那我应该相信哪个结果呢?GPC是相对较正的结果,我觉得标样的这点差异不至于对结果有那么大的影响吧?而且阿我们每次购买标样都不可能是与上次分子量相同的,这样的数据不是没多大的可比性?请求各位帮忙解惑啊!在下先感激了!
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分流管线、检测器、注射器的清洗 1.分流管线的清洗: [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]用于有机物和高分子化合物的分析时,许多有机物的凝固点较低,样品从气化室经过分流管线放空的过程中,部分有机物在分流管线凝固。 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]经过长时间的使用后,分流管线的内径逐渐变小,甚至完全被堵塞。分流管线被堵塞后,仪器进样口显示压力异常,峰形变差,分析结果异常。在检修过程中,无论事先能否判断分流管线有无堵塞现象,都需要对分流管线进行清洗。分流管线的清洗一般选择丙酮、甲苯等有机溶剂,对堵塞严重的分流管线有时用单纯清洗的方法很难清洗干净,需要采取一些其他辅助的机械方法来完成。可以选取粗细合适的钢丝对分流管线进行简单的疏通,然后再用丙酮、甲苯等有机溶剂进行清洗。由于事先不容易对分流部分的情况作出准确判断,对手动分流的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]来说,在检修过程中对分流管线进行清洗是十分必要的。 对于EPC控制分流的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],由于长时间使用,有可能使一些细小的进样垫屑进入EPC与气体管线接口处,随时可能对EPC部分造成堵塞或造成进样口压力变化。所以每次检修过程尽量对仪器EPC部分进行检查,并用甲苯、丙酮等有机溶剂进行清洗,然后烘干处理。 由于进样等原因,进样口的外部随时可能会形成部分有机物凝结,可用脱脂棉蘸取丙酮、甲苯等有机物对进样口进行初步的擦拭,然后对擦不掉的有机物先用机械方法去除,注意在去除凝固有机物的过程中一定要小心操作,不要对仪器部件造成损伤。将凝固的有机物去除后,然后用有机溶剂对仪器部件进行仔细擦拭。 2、TCD和F ID检测器的清洗 TCD检测器在使用过程中可能会被柱流出的沉积物或样品中夹带的其他物质所污染。TCD检测器一旦被污染,仪器的基线出现抖动、噪声增加。有必要对检测器进行清洗。 HP的TCD检测器可以采用热清洗的方法,具体方法如下: 关闭检测器,把柱子从检测器接头上拆下,把柱箱内检测器的接头用死堵堵死,将参考气的流量设置到20 ~ 30 ml/min, 设置检测器温度为400℃,热清洗4~8 h,降温后即可使用。 国产或日产TCD检测器污染可用以下方法。仪器停机后,将TCD的气路进口拆下,用50 ml注射器依次将丙酮(或甲苯,可根据样品的化学性质选用不同的溶剂)无水乙醇、蒸馏水从进气口反复注入5~10次, 用吸耳球从进气口处缓慢吹气, 吹出杂质和残余液体, 然后重新安装好进气接头, 开机后将柱温升到200 ℃, 检测器温度升到250 ℃, 通入比分析操作气流大1~2倍的载气, 直到基线稳定为止。 对于严重污染, 可将出气口用死堵堵死, 从进气口注满丙酮(或甲苯,可根据样品的化学性质选用不同的溶剂) ,保持8 h左右,排出废液,然后按上述方法处理。 FID检测器的清洗: F ID检测器在使用中稳定性好,对使用要求相对较低,使用普遍,但在长时间使用过程中,容易出现检测器喷嘴和收集极积炭等问题,或有机物在喷嘴或收集极处沉积等情况。对FID积炭或有机物沉积等问题,可以先对检测器喷嘴和收集极用丙酮、甲苯、甲醇等有机溶剂进行清洗。当积炭较厚不能清洗干净的时候,可以对检测器积炭较厚的部分用细砂纸小心打磨。注意在打磨过程中不要对检测器造成损伤。初步打磨完成后,对污染部分进一步用软布进行擦拭,再用有机溶剂zui后进行清洗,一般即可消除。 3、注射器的清洗 注射器使用前可先用丙酮清洗,以免玷污样品,但更好的方法还是用待注射样品对注射器本身做一二次清洗。清洗时只能吸入样品,排出样品时要在样品瓶之外。注射器在使用结束后要立即清洗,以免被样品中的高沸点物质玷污。一般常用下述溶液依次清洗:5%氢氧化钠水溶液、蒸馏水、丙酮,zui后用真空泵抽干。 用于有机物和高分子化合物定量分析的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]一般采用分流进样,毛细管色谱柱。根据仪器的生产厂家和型号的不同,进样口的分流控制系统一般有EPC控制分流和手动控制分流两种情况。 在检修时,对[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]进样口的玻璃衬管、分流平板,进样口的分流管线, EPC等部件分别进行清洗是十分必要的。 玻璃衬管和分流平板的清洗: 从仪器中小心取出玻璃衬管,用镊子或其他小工具小心移去衬管内的玻璃毛和其它杂质,移取过程不要划伤衬管表面。 如果条件允许,可将初步清理过的玻璃衬管在有机溶剂中用超声波进行清洗,烘干后使用。也可以用丙酮、甲苯等有机溶剂直接清洗,清洗完成后经过干燥即可使用。 分流平板zui为理想的清洗方法是在溶剂中超声处理,烘干后使用。也可以选择合适的有机溶剂清洗:从进样口取出分流平板后,首先采用甲苯等惰性溶剂清洗,再用甲醇等醇类溶剂进行清洗,烘干后使用。
参加实验室比对,A和B的检测结果不同,怎么处理?A检出,B未检出。然后应该怎么处理?需要内审、仪器期间核查等等么?
[font=宋体]发帖人:[/font]carollee[font=宋体]链接:[/font][url=https://bbs.instrument.com.cn/topic/5846830][color=windowtext]https://bbs.instrument.com.cn/topic/5846830[/color][/url][b][font=宋体]问题描述:[/font][/b][font=宋体]分流进样是毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的首选的进样方式,适用于大部分气体或液体样品的分析,尤其对未知样品或已知[/font]“[font=宋体]较脏[/font]”[font=宋体]的样品,使用分流进样,可保护毛细管柱,减小或避免色谱柱被污染的可能。该进样方式进样时,进入进样口的样品大部分被分流掉而没经过色谱柱,仅有少量的样品进入毛细管柱。分流比就是分流流量与柱流量的比值。例如,如果进入色谱柱的流量就是[/font]1 mL/min[font=宋体],分流流量为[/font]20 mL/min[font=宋体],那么分流比是[/font]20[font=宋体]︰[/font]1[font=宋体]。[/font][font=宋体]实际工作中,我们应该如何测定分流比和分流量[/font]?[font=宋体]分流比对物质的分离测定又有何影响[/font]?
就是比如两个不同分析人员做比对比对报告要包括哪些统计检验呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP