椭偏仪结构原理与发展[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=63165]椭偏仪结构原理与发展[/url]
各位好,请问哪里有椭偏仪可以提供测试?最好是在广州的
我们实验室有一台德国进口的椭偏仪,型号为:SpecEI-2000-VIS,测量时发现同一个点测量时结果都会有偏差。几次图谱的拟合曲线不一致。有谁知道的告诉小弟一声。非常感谢。
求教大神,这是老师给的任务 用椭偏仪测薄膜反射率 求教一下 怎么测?
[em06] 这几种光谱型椭偏仪优势差异在哪?????SENTECH椭偏仪,HJY椭偏仪,Sopra S.A.,AQUILA公司
要做毕业设计,有关椭偏仪和椭偏算法的,不知道在哪里可以找到相关的资料,望大家指教指教!~
供大家参考![img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=55565]椭偏仪测试聚合物膜厚[/url]
最近工作上涉及到椭偏仪,想知道椭偏仪测量的优点与缺点都有什么?我看了一些资料,说椭偏仪测量存在如下缺点:1、薄膜基片如果是透明的,那么对样品的前处理会很麻烦,很费钱2、如果样品是有机物薄膜,椭偏仪很难测出结果来3、椭偏仪测量得到的数据需要手工处理,很复杂,非专业人员无法处理,暂时没有软件可以直接得薄膜的厚度、折射率、吸收系数值4、测量吸收系数时不能同时测量透射光与反射光,所以得到的吸收系数是有误差的不知道以上观点是不是正确? 为什么薄膜的基片不能是透明的?透明的基片对薄膜厚度测量有什么影响?为什么很难得到有机物薄膜的厚度?请指教,谢谢!不知道是否还有其他缺点?
在si片上镀一层纳米碳膜,碳膜表面光洁如镜面,采用椭偏仪测量薄膜的厚度,测量时选择基底材料为si,将k设为0(透明薄膜,不知是否准确或透明薄膜如何定义?),根据SEM测量得到的薄膜厚度拟合得到一个n值(2.0921),采用此n值对类似情况下制备的薄膜进行厚度测量,测量得到的结果还行,基本与肉眼看到的薄膜厚度差别相当。不知此方法是否正确?1.是否能采用透明膜的测量方法测量碳膜?2.采用同一n值测量厚度是否合适?希望高手指点,谢谢!
检测薄板上油膜厚度的仪器,我们目前使用的是日本岛津公司生产的AEP-300椭圆仪(非椭偏仪),但已经停止生产。仪器使用多年故障逐渐增多。美国DONART公司有类似仪器,但是单片检测,不适合用于大生产。请问专家,还有没有同类仪器可供选择?

各位好,请问SENTECH SE400adv 椭偏仪在同一个点测试时,N值会出现偏差,,,是怎么回事,怎么可以解决好基准是不是不可以超过0.002http://ng1.17img.cn/bbsfiles/images/2016/08/201608181737_605416_3135057_3.png
扫描电镜可以测反射率?还是椭偏仪可以?
请问各位大虾,我是HPLC的菜鸟,现在正在尝试测植物叶片的有机酸,可是不知为什么做重复时各峰的延迟时间一般会有少许提前呀,这样就无法正确的定性了?各位有什么好的意见吗,我用的是pH1.8的NH2HPO4缓冲液,先谢了:)
在试方法过程中,溶剂延迟的时间短,让溶剂出峰了,且目标物在溶剂前出峰啦,这该怎么办呢?求助求助。拜托拜托

http://ng1.17img.cn/bbsfiles/images/2017/03/201703271448_01_2911392_3.jpg如图:有微友提问道:各位大神,一个标准系列,为啥有的洗脱时间延迟呢?有微友回复道:这是柱效降低了吗?各位大神、大伽纷纷过来探讨,看看这是啥原因呢?http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif
今天看到一则解释气-质联用如何设置溶剂延迟时间的帖子: 如下一般溶剂延迟的时间设置在溶剂峰出来之后,待测分析组分的前面。只要溶剂出峰时,保证灯丝关闭即可,灯丝就不会烧坏或影响寿命。如果前一段时间不用采样,也可以把延时设置到此时间后。 如果有GC,在与GCMS相同的条件下,进一针溶剂,测定溶剂的出峰时间。 顶空进一点所用的溶剂,测定溶剂的出峰时间。 大分流比下,针点进一点溶剂,测定溶剂的出峰时间。气质连用是否会出现这样一种情况,待测组分先出峰,溶剂后出峰,如果出现这样一种情况,该如何设置溶剂延迟时间?
我用ECD检测器和质谱联用,乙酸乙酯作溶剂,溶剂延迟设置为3min,可是ECD检测器上也不出峰,这是怎么回事?溶剂延迟不只是质谱灯丝的开关延迟时间吗?对ECD检测器应该没有影响吧
今年年底CMA证书到期,又准备到年底搬迁,时间上有冲突,不知可否申请延迟评审?如果可以延迟时间有没具体规定?
客户方法转移中进样量200μl,延迟体积设置为50μl,保留时间为17.5min,而我方样品环为100μl,上样量100μl,不设置延迟体积,保留时间为16.5min,请问,怎样设置才能使我方出峰时间为17.5min,按理说换大的样品环才会增加延迟体积吧
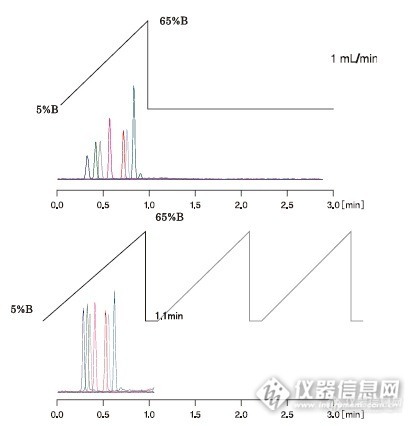
在“液相色谱”论坛中看见有网友在讨论延迟体积是什么?并且有些什么影响?在这里,就我的理解给大家一个参考。延迟体积主要是指从泵出来之后到进入色谱柱之前这一段流路的体积,这一段流路中最大的体积来自于混合器,混合器的大小主要决定了延迟体积的大小。那么,问题来了,这部分体积对分析的影响表现在什么地方呢?如同“zyulcl”所说,对于等度分析而言是没有影响的,但在梯度分析时影响就能显现出来。首先,我们来看下图,这是理论梯度和真实梯度的区别,也叫梯度追随性。因为延迟体积的存在,我们可以设想为流动相在这部分体积中会进行一个再混合的过程(类似于样品在塌陷的柱头处扩散),这样真实梯度就会表现为梯度按曲线变化(如上部的色谱图);当仪器的延迟体积变小时,真实梯度会逐渐向理论梯度靠近(如下部的色谱图)。http://ng1.17img.cn/bbsfiles/images/2015/06/201506241102_551267_2222981_3.jpg那么,这种梯度追随性的差异会对分析工作带来什么影响呢?首先,因不同仪器的延迟体积不同,导致相同梯度设定下,达到某一特定混合比例的时间不同(在X仪器上达到A:B=40:60,在3min;在Y仪器上达到A:B=40:60,在3.2min)。其次,拿上图举例,上部色谱图显示的仪器,达到90%B相的时间段非常短;下部色谱图显示的仪器,达到90%B相的时间段就更接近于理论梯度。对于这一极性流动相下样品的洗脱时间就更长。最后,我们看下图,一般在梯度分析的末尾,我们都需要把流动相切换成梯度起始时的配比,而延迟体积更大的仪器,需要更多的回冲时间,从而影响分析效率。同理,延迟体积小的仪器更能提高分析效率,这一点在以3-5min为一分析周期的LCMS分析中更加明显。http://ng1.17img.cn/bbsfiles/images/2015/06/201506241122_551277_2222981_3.jpg以上是我关于延迟体积对液相分析影响的一些认识,欢迎大家斧正和讨论。
不是设定溶剂延迟是为了保护灯丝吗?为什么每次运行时,仪器都会自动跳出警告:溶剂延迟,会缩短灯丝寿命
我现在需要测量ITO薄膜的厚度,透明的薄膜,大概200纳米左右。用椭偏仪行不行?是不是还需要薄膜的折射率?希望各位大侠指导一下[em61] [em09]
用酸性铬兰K-萘酚绿B作指示剂,用5毫升1比1三乙醇胺,0.5mlKCN作屏蔽剂,氨水-氯化铵作缓冲剂,EDTA滴定,颜色变化由红色-紫色-蓝色,紫色变蓝色过渡时很缓慢,延迟。怎么解决?蓝色也不太好观察
[color=#444444]目前用的岛津的lc-20at系列。[/color][color=#444444]自己现在摸索梯度洗脱,发现梯度的时候有很大的延迟,比如1-17min水的浓度由50%-60%,那么基线大概就会在11分钟左右就开始下降,也就是说有10min左右的延迟,不知道我说得对不对。如果我的出峰时间大概在15min左右,那么我是不是就要提前10分钟,也就是在5min左右开始梯度的变化。[/color]

前 言:在许多型号的原子吸收分光光度计的条件设置中,有一项称为【延迟时间】参数。关于此项参数的功能和设置,往往有许多仪器使用者不甚了解。图-1,图-2是两款原吸仪器关于延迟时间设定的操作界面:http://ng1.17img.cn/bbsfiles/images/2011/08/201108112250_309907_1602290_3.jpg图-1 火焰方式下的延迟时间的设定界面http://ng1.17img.cn/bbsfiles/images/2011/08/201108112251_309908_1602290_3.jpg图-2 火焰方式下的延迟时间的设定界面(一) 火焰测量方式下的延迟时间的作用和设置:上面两张图例列出的就是仪器分析方法的参数设定项目,其中就有【延迟时间】一项(注:红色框内)。这个时间的设置就是从仪器启动测量开始起到仪器开始计算的间隔时间。众所周知,火焰分析的计算一般是指对样品信号的测量面积的积分计算,这个被积分的面积一般为矩形。可是样品从进样开始起到形成稳定的测试信号需要一个上升的时间过程,也就是说样品测试信号的的形成有一个上升的斜率,如图-3显示:http://ng1.17img.cn/bbsfiles/images/2011/08/201108112252_309909_1602290_3.jpg图-3 1ppm浓度的铜标准溶液的测试曲线(上升时间5秒)这个斜率上升所需要的时间,就是设定延迟时间长短的依据。但是不同的样品,不同的浓度其粘稠度也不一样,因此这个斜率没有规律可言,这就给延迟时间的设定造成了一定的难度。如图-4和图-5所示:http://ng1.17img.cn/bbsfiles/images/2011/08/201108112332_309919_1602290_3.jpg图-4 10ppm浓度的铜标准溶液的测试曲线(上升时间7秒)http://ng1.17img.cn/bbsfiles/images/2011/08/201108112255_309912_1602290_3.jpg图-5 20ppm浓度的铜标准溶液的测试曲线(上升时间8秒)
现在使用的是安捷伦1260的馏分收集器,制备的时候不接检测器,直接根据时间段来回收馏分。请问大家,这种情况还需要设置延迟校正的体积吗,我没明白延迟校正是校正的从检测器到馏分收集器的管路体积还是检测上的延迟啊
谁能帮忙解释一下“溶剂延迟”,如何计算“溶剂延迟?
agilent的延迟体积怎么计算。agilent1100的延迟体积是多少?
如题,就是一层薄膜,如果用椭偏仪的话是不是一定要知道基底的折射率阿,可是这个也是不知道的,请问还有其他办法吗?
科研故事:常被轻视的方法研究2012-04-22 22:14 来源:科学网 作者:徐 耀 科研浮躁的一个重要表现是,只重视实验结果(或者数据),而对获得这些结果(或者数据)的方法重视不够,所以经常对别人发表的好结果望洋兴叹,而不去下辛苦掌握些方法论。举个例子,给定一层厚度在100-200纳米的有支撑透明光学薄膜,如何准确测量其厚度和光学参数(比如最根本的折射率和消光系数)?猛一看,这似乎根本不是个问题,甚至相关的商用仪器都有很多,但深入分析一下,事情就没有那么简单。原则上,厚度和折射率、消光系数等参数均可通过椭圆偏振仪(椭偏仪)一次性获得,如果是光谱扫描型椭偏仪,则薄膜的色散曲线都可以获得。但是椭偏仪测量基于物理模型,这些模型因物质而不同,对于实际工作中千变万化的薄膜材料和结构,要么物质特殊,要么结构特殊,只要不符合仪器的模型库,测量就无法进行,当然也可以自己建立物理模型,但这个就不是购买仪器的初衷了。所以,不能指望商用仪器解决一切,还是要自己掌握其中的方法论。还是这个光学薄膜,我们建立了自己的方法论。首先,通过X射线反射谱准确测量薄膜的绝对物理厚度,这是不依赖于任何物理模型的,是由薄膜上下表面反射光干涉引起的,只要量出反射率曲线中相邻震荡峰的间隔就可以获得薄膜厚度。然后,以这个厚度为初始参数,对薄膜的透射谱进行拟合,以获得薄膜的光学参数。基于分光法的透射谱很准确,而透明基底会给椭偏仪测量带来额外误差,因此我认为对透射谱进行拟合要比依靠椭偏仪靠谱。利用透射谱拟合也要涉及几种色散模型,但这些模型是通用模型,与薄膜结构无关,经过对比研究后应该可以确定最适合的模型。在此基础上,我们可以对不同类型的薄膜区别对待,建立各自的测量方法。我们以前认为只要有了椭偏仪,薄膜测量就没问题了,没想到有了仪器,问题更多更让人烦恼,因为得到的数据常常超出正常。我们做的这些工作,不具备太多创新性,按理说不符合现在对科研的期待,但是我们掌握了方法,我认为这样的工作很有意义,一旦方法建立,就像给火车铺设了轨道。因此,仪器重要,方法论是发挥仪器作用的保障。回过头来,我们为什么经常轻视甚至忽视方法论的研究呢?我认为原因有三。第一,科研考核注重结果的显示度,科研管理偏于目标管理,而非过程管理,科研人员只盯着最终数据。第二,科研人员眼界狭窄,方法论研究往往要深入理论,而理论是很多实验研究人员不愿意碰的。第三,谁都不是哪吒三头六臂,但科研合作不足使方法论研究难以开展。我国目前花大量外汇购买的高精尖仪器很多了,但用得足、用得好的并不多,以至于功能闲置,造成极大浪费,其中对方法论的掌握不足大概是主要原因。因此,科研人员应该多关注方法论研究。







 我要推广仪器
我要推广仪器
 下载APP
下载APP