请问大家谁做过苯乙胺的HPLC检测?为什么非得衍生化呢?http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
[color=#444444]DB-624色谱柱,按照限度配制二乙胺溶液出峰有些拖尾,做其线性的时候,25%和50%的浓度出峰均出现漂移(出峰时间推后)的现象,这样定量限就没办法做,出峰时间和原来的相差0.3min左右。稀释溶剂DMF,出峰均正常。请问有可能什么原因,导致二乙胺出峰异常,仪器:安捷伦7697A/7890B[/color]
最近需要检测酯类物质,其中含有1-4PPm的三乙胺,每次检测,都是同一个样品三乙胺含量忽高忽低,有时能达到几十PPm,我的仪器条件是福立9790二型,柱温起始80,保持2分钟,然后20度/min至200,检测器250,进样口230,样品4各组分,乙醇,苯,三乙胺,酯类,色谱柱是DB-5,想请大侠帮帮忙,有什么好办法没有?是三乙胺这种物质在色谱中残留呢,还是气化不好?
求三乙胺溶剂残留气相检测方法
大家好!我是医药单位质量分析室的一名研究人员,针对我公司产品的工艺改进,需要对反应后的母液进行分析检测,母液里面含甲醇、二乙胺、苯甲醛、水等物质,现在领导要求建立合适的方法,对母液的质量进行监控。我想请问大家,能不能运用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]方法进行啊?针对这几种成分,选择什么样的检测器和色谱柱好呢?由于任务急,希望大家能给点建议,不甚感激!!!
样品偏酸性,要检测里面的三乙胺溶剂残留,请各位给个好一些的气相检测方法,谢谢!
建立一种简单、快速、准确同时测定大米蛋白粉中三聚氰胺及其类似物三聚氰酸、三聚氰胺一酰胺、三聚氰胺二酰胺的方法。样品经二乙胺- 水- 乙腈(1:4:5,V/V)超声提取30min 后,提取液于70℃条件下氮气吹干,在吡啶介质中经甲基硅烷化衍生后采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]- 质谱仪测定(内标法定量)。此方法检测限为0.5mg/kg、线性相关系数(20~1000μg/L 范围内)不小于0.9989;当加标量为1.0~5.0mg/kg 时,平均回收率为72.01%~96.45%、相对标准偏差不大于6.3%。此方法可用于大米蛋白粉中三聚氰酸及其类似物的检测。
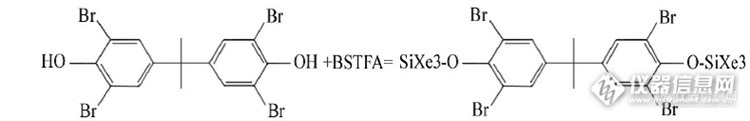
羧酸。此外,还用到双乙烯酮(见乙烯酮)和羧酸酯等酰化可分为以下三种主要类型①C-酰化 是一种形成新的碳-碳键的缩合反应,其中最重要的是酰基取代芳环上的氢生成芳酮的过程。例如醋酐与萘在二氯乙烷溶剂中,在90°C发生如下酰化反应:②N-酰化 伯胺基或仲胺基上的氢被酰基取代生成酰胺的过程应通式: (RCO)2O + R`NH2 → R`NHCOR + RCOOH③O-酰化 醇或酚分子中羟基的氢原子被酰基取代或炔烃与酸反应的过程,因为生成的产物是酯,所以又称酯化常用的酰基化试剂有:乙酸酐、三氟乙酸酐、五氟丙酸酐、七氟丁酸酐、N-甲基二(三氟乙酰胺)、1-(三氟乙酰基)咪唑等。3)烷基化是烷基由一个分子转移到另一个分子的过程。是化合物分子中引入烷基(甲基、乙基等)的反应。如汞在微生物作用下在底质下会烷基化生成甲基汞或二甲基汞。工业上常用的烷基化剂有烯烃、卤烷、硫酸烷酯等①烷烃的烷基化,如用异丁烯使异丁烷烷基化得高辛烷值汽油组②芳烃的烷基化,如用乙烯使苯烷基化http://ng1.17img.cn/bbsfiles/images/2014/09/201409251121_515546_1738073_3.jpg③酚类的烷基化,如用异丁烯使对甲酚烷基化常用的烷基化试剂有重氮甲烷等4)硅烷化通过硅烷基取代羟基上的活性氢进行的(气相/气质使用最多)硅烷是一类含硅基的有机/无机杂化物,其基本分子式为:R'(CH2)nSi(OR)3。其中OR是可水解的基团,R'是有机官能团如:BSTFA 与TBBP-A 的衍生化反应是通过硅烷基取代TBBPA 羟基上的活性氢进行的。衍生反应的产物是硅醚(我这次要检测的物质,衍生试剂为BSTFA+TMCS 99:1)http://ng1.17img.cn/bbsfiles/images/2014/09/201409251124_515547_1738073_3.pngTMCS本身也是硅烷化试剂,只是硅烷化较弱,加入BSTFA中主要是作为催化剂。衍生化温度大致70度左右。在衍生过程中加入稍过量的衍生化试剂可减少容器表面的吸附和可能存在的少量水常用的硅烷化试剂有:1、N,O-双三甲基硅基三氟乙酰胺(BSTFA)和N,O-双三甲基硅基乙酰胺(BTA):能衍生化胺基和羟基。常用于体内药物及其代谢物的检测。 2、N-甲基叔丁基二甲基硅基三氟乙酰胺(MTBSTFA):常用于药物、类固醇的检测。由于叔丁基二甲基硅基有较大的空间效应,有些胺基较难衍生化。在质谱检测中,该衍生物很容易失去叔丁基形成较强的(M-57)+离子,有利于作为MS-MS的母离子。 3、N-甲基三甲基硅基三氟乙酰胺(MSTFA):最常用的硅烷化试剂之一。衍生化也是引入三甲基硅基4.衍生化对气质的作用1)GC-MS方法分析样品时,对羟基、胺基、羧基等官能团进行衍生化有十分重要的作用,主要表现在a、改善样品的气相色谱性质。如羟基、
聚山梨酯80残留溶剂检测中的(三乙胺与二甲基甲酰胺),三乙胺的出峰面积越来越大。不知道大家有没有碰到过这种情况,出峰面积一针比一针大,求指点!
最近在做水质三乙胺的检测,发现校准曲线很难做直,请问主要有哪些影响因素,还有三乙胺的标样何处可以买到?
??液相色谱使用最多的是紫外检测器,为了使一些没有紫外吸收或紫外吸收很弱的化合物能被紫外检测器检测,往往通过衍生化反应在这些化合物的分子中引入有强紫外吸收的基团:2,4-二硝基苯、苯甲基、对硝基苯甲基、3,5-二硝基苯甲基、苯甲酸酯、对甲苯酰、对氯苯甲酸制、对硝基苯甲酸酯、对甲氧基苯甲酸酯、苯甲酰甲基、对溴苯甲酰甲基,这些衍生物可被紫外检测器检测。??大多数紫外衍生反应来自经典的光度分析和有机定量分析,新的衍生化反应和衍生化试剂是随液相发展而发展的,这些反应的原理都来自有机合成,所以就要求操作者对有机合成有所了解。但是,由于柱前衍生化是为色谱分析准备样品,处理样品的量小(mg级),所用的小型和微型反应器皿又不同于常量的有机合成,而是类似于近年来发展的微量有机合成。紫外衍生化反应要选择反应产率高、重复性好的反应。过量试剂和试剂中的杂质如果干扰下一步的色谱分离和检测,则在色谱进样前要进行纯化分离。还要注意反应介质对紫外吸收的影响。??(1)苯甲酰化反应:苯甲酰氯及其衍生物--对硝基苯甲酰氯、3,5-二甲基苯甲酰氯和对甲氧基苯甲酰氯都可以同胺、醇和酚类化合物反应,生成紫外吸收的苯甲酸酯类衍生物;??(2)2,4-二硝基氟代苯(DNFB)的反应:DNFB与醇的反应产率很低,但可与大多数伯胺、仲胺和氨基酸反应,生成强紫外吸收的苯胺类衍生物;??(3)苯基异硫氰酸酯(PITC)的反应:PITC可与氨基酸反应,生成苯基己丙酰硫脲衍生物--PTH氨基酸;??(4)苯基磺酰氯的反应:苯基磺酰氯可与伯胺和仲胺反应。??甲苯磺酰氯可与多氨基化合物反应,不仅能提高它们的检测灵敏度,还可改变HPLC的分离度。??(5)有机酸的酯化反应:有机酸很容易与酰溴基反应生成酯,常用的酰溴基试剂有苯甲酰溴、甲氧基苯甲酰溴、对溴基苯甲酰溴和对硝基苯甲酰溴等;??酯化反应在极性溶剂(如乙腈、丙酮或四氢呋喃)中进行,有时需要加催化剂,如冠醚加钾离子、二乙胺或N-二异丙基胺等。??(6)羰羟基化合物的反应:醛类和酮类中的羰基可与2,4-二硝基苯肼(DNPA)反应,反应在弱酸性条件下进行,生成苯腙衍生物。??羰基化合物还可以与对甲基苄基羟胺(PNBA)在碱催化下反应,生成有强紫外吸收的肟。
三乙胺的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测方法,请赐教
最近采用顶空做某头孢药物中三乙胺检测,总有拖尾峰出现,效果不理想;后来我自己手动进样,还是有些拖尾,但不是很严重;查找资料后,拖尾峰的原因很多。是不是我加了NaOH的缘故呢?还有,做其他都还不错的。各位,帮我分析分析哈~~
请大家帮提下宝贵建议我使用的仪器是安捷伦的7890A-5975C气质联用另外配有FID检测器,使用顶空进样的方法。在检测某些烟标按照国家标准检测的条件苯峰处总是会有三乙胺干扰苯峰,峰型是个拖尾峰,已致无法对其进行准确定量。三乙胺是油墨中引入的。由于烟标VOCs检测国家标准没有使用质谱,无法对其进行定性,所以很多时候会对该项引起误判,在国标方法的前提下,请问大家有解决的办法吗?
GB 14680-93 标准中3.2吸收液的配制 ,配制二硫化碳吸收液的时候,需要对二乙胺和三乙醇胺的试剂进行新蒸馏提纯,但标准中并没有具体提及到提纯的方法,如试剂不提纯会影响吸收液的颜色,这样会对检测结果造成偏差,现想咨询一下各路大神,是否有具体的提纯方法,谢谢
各位前辈,小弟想问一下乙二醇甲醚和三乙胺能用液相色谱分析吗,如果能流动相是什么啊?条件呢?我们的液相是带紫外的检测器。谢谢
样品需要衍生后才能进样,衍生过程中添加了10ul0.5mol/L三乙胺,反应结束用等量盐酸中和,然后样品稀释了10倍,采用负离子监测,请问这能直接进质谱吗?
用GC-MS对三聚氰胺进行检测时,需要采用BSTFA+TMCS对三聚氰胺进行衍生化处理后再分析。我不确定自己是否成功进行衍生化反应了,想知道衍生后的产品名称,最好能提供CAS号或者结构式,万分感谢!着急啊,试验老是做出来感觉不对。。。。
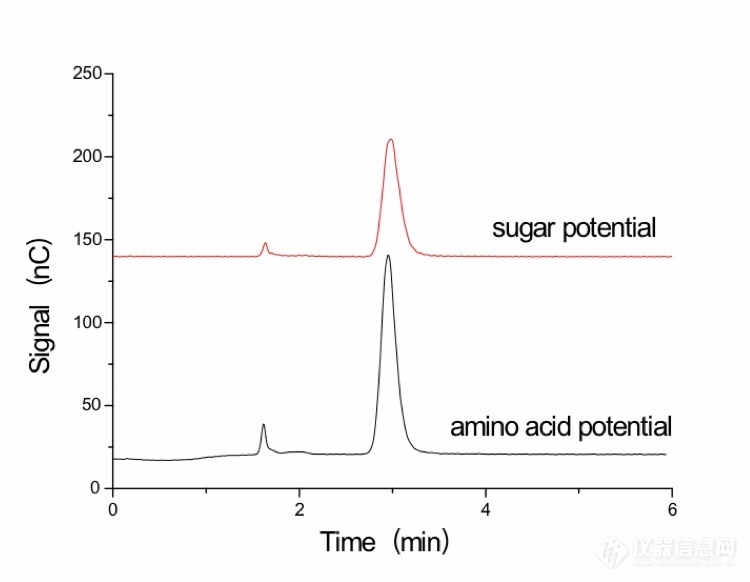
[align=center][b][url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱后补液-积分脉冲安培法检测阿立哌唑中残留三乙胺[/b][/align][b]摘要[/b]目的:建立测定阿立哌唑原料药中三乙胺残留量的新方法。方法:采用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱后补液-积分脉冲安培法。采用阳离子交换柱,以30 mmol/L甲烷磺酸为淋洗液,流速1.0 mL/min;柱后补液为500 mmol/L NaOH溶液,流速0.2 mL/min;波形为氨基酸电位。结果:三乙胺在0.1322-1.322 mg/L范围内线性关系良好(R[sup]2[/sup]=0.9994),加标回收率在101.7%~105.9%之间,RSD为1.85%(n=6)。结论:建立的方法准确、可靠、灵敏度高,适用于测定阿立哌唑原料药中三乙胺的残留量分析。[b]关键词[/b][url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url];积分脉冲安培;阿立哌唑;三乙胺阿立哌唑(aripiprazole),化学名为7-{4--丁氧基}-3,4-二氢-2(1H)喹啉酮,是日本Otsuka公司开发的新型非典型抗精神病药,临床主治各种急、慢性精神分裂症和情感障碍[sup][/sup]。阿立哌唑的合成路线较多[sup][/sup],在合成过程中曾用到三乙胺,因此产品中有可能会残留微量的基因毒性杂质三乙胺,由于三乙胺具有助溶和轻度的防腐作用,因此对三乙胺残留量的监测是阿立哌唑药物质量控制过程中必不可少的一部分。目前三乙胺已收载于人用药品注册技术要求国际协调会(ICH),Q3C(R6)中[sup][/sup],规定其限度为500mg/Kg,欧洲药品质量管理局(EDQM)也规定其残留限度为320mg/Kg。通常,三乙胺采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]和溴酚蓝分光光度法[sup][/sup]进行测定。有不少人采用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]检测乙醇胺、二甲胺等胺类[sup][/sup],但用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]测定三乙胺很少见。李向春[sup][/sup]等采用Dionex IonPac CS17色谱柱,MSA 6mM等度淋洗,采用CSR循环再生电抑制模式测定了草甘膦合成工艺中的三乙胺。上述方法中检出限最低的为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,可达约48 mg/kg。但阿立哌唑中残留的三乙胺含量很低,采用上述方法灵敏度不够,且阿立哌唑中主体干扰较大,无法满足要求。潘思[sup][/sup]等采用柱后衍生[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]-脉冲安培法来测定盐酸羟胺,采用Dionex IonPac CS16 (4×250 mm)色谱柱,流动相是30 mmol/L甲磺酸溶液,流速为1.0 ml/min;衍生剂为500 mmol/L氢氧化钠,流速为0.3 ml/min,该方法检测限为0.012 mgL[sup]-1[/sup],定量限为0.037 mgL[sup]-1[/sup]。因此本文借鉴该方法来测定三乙胺的含量,并对淋洗液浓度及流速、柱后补液的浓度及流速、电位波形进行优化,通过考察其线性关系、精密度、稳定性来验证方法的可行性。[b]1实验部分1.1 仪器与试剂[/b]Thermo ICS5000+ 型[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url],包括单元四元梯度泵,AS-AP自动进样器,DC模块含安培检测器,Chromeleon 6.80色谱工作站;色谱柱为Dionex IonPac CS17 (4×250 mm),保护柱为Dionex IonPac CG17(4×50 mm); Mettler Toledo AL204型电子分析天平;Millipore-Q Advantage A10型超纯水机。三乙胺(99.5%),盘锦研峰科技有限公司;甲烷磺酸(99.5%),阿拉丁试剂有限公司;50% NaOH(w/w),分析纯,德国Merck公司; OnGuard[sup]TM [/sup]RP柱(1 cc),美国Thermo公司;阿立哌唑供试品(1[sup]#[/sup],2[sup]#[/sup],3[sup]#[/sup]),某药厂提供。[b]1.2 溶液的配制[/b]1.2.1 三乙胺标准溶液的配制精确称取66.1 mg三乙胺标准品于50 mL容量瓶中,用30 mmol/L MSA淋洗液溶解定容,配制得到132 2 mg/L标准储备液,稀释得到26.44 mg/L的标准溶液。再逐级用淋洗液稀释得到浓度分别为1.322 mg/L、0.661 0 mg/L、0.440 7 mg/L、0.264 4 mg/L、0.132 2 mg/L的三乙胺系列储备液。1.2.2 甲烷磺酸淋洗液溶液的配制称取19.28 g 甲烷磺酸于2.0 L PP淋洗液瓶中,加超纯水到2000 mL,摇匀,所得溶液的浓度约为100 mmol/L。1.2.3 NaOH溶液的配制称取81.09 g 50%NaOH(w/w)于2.0 L PP淋洗液瓶中,加超纯水到2000 mL,摇匀,所得溶液的浓度约为500 mmol/L。1.2.4 实际样品溶液的配制称取50.1 mg 阿立哌唑供试品于25 mL容量瓶中,准确加入10 mL乙腈,置于50℃水浴中溶解,摇匀,随后准确加入10 mL超纯水,置于冰箱中冷藏60分钟后取出,阿立哌唑会以沉淀形式析出。静置后离心,取上清液用经活化的RP柱(活化方式:先用5 mL甲醇对RP柱进行冲洗,放置30 min后,用10 mL超纯水进行冲洗,备用)进行过滤,先丢弃最初的3 mL,取滤液即得浓度约为2500 mg/L的供试品溶液。[b]1.3 色谱条件[/b]淋洗液:A 超纯水(70%),B 100 mmol/L甲烷磺酸(30%),流速为1.0 mL/min,等度淋洗;柱后补液:500 mmol/L NaOH溶液,流速为0.2 mL/min;波形:氨基酸电位;进样量:25 μL;柱后衍生管:375μL。[b]2 结果与讨论2.1 色谱条件的确定[/b]2.1.1 淋洗液浓度的选择实验选取20 mmol/L、25 mmol/L、30 mmol/L的甲烷磺酸溶液作为淋洗液分别测定1.322 mg/L的三乙胺标样,检测结果显示其保留时间分别为3.567min、3.189min、2.953min,峰高分别为47.31 nC、103.7 nC、119.8 nC。表明30 mmol/L的淋洗液灵敏度最高,且保留时间适宜。若使用更高浓度的甲烷磺酸溶液作为淋洗液,则三乙胺的保留时间会更短,但可能存在与其他快出峰杂质分离度变差,影响定量。因此选取30 mmol/L的甲烷磺酸溶液作为实验的淋洗液。2.1.2 检测电位的选择伯胺的有机化合物,在碱性条件下,用金电极采用糖电位和氨基酸电位都有较高的响应。糖电位为脉冲安培检测,氨基酸电位为积分安培检测,脉冲安培检测在一个脉冲周期中对电流积分所施加的电位是单一的,它存在一个短暂的间歇以使充电电流衰减,而积分安培对工作电极施加的是一种对应时间波形的循环电位,即电极先被氧化然后再被还原为其原始状态。因此,在CS17柱分离后,用NaOH补液调节pH到碱性。选取糖电位和氨基酸电位这两种波形分别测定1.322 mg/L的三乙胺标样,结果如图1。从图1可以看出,二者的噪音差别不大,且氨基酸电位波形的响应值高,因此选取氨基酸电位波形作为实验的波形。[align=center][img=,690,535]https://ng1.17img.cn/bbsfiles/images/2019/08/201908312150078580_1354_3426139_3.jpg!w690x535.jpg[/img][/align][align=center][b]图1 电位波形的影响[/b][/align][align=center]Fig.1 Effect of potential waveform[/align]2.1.3 柱后补液流速的选择实验选取氨基酸电位波形的柱后补碱NaOH溶液的0.2 mL/min、0.3 mL/min流速分别测定1.322 mg/L的三乙胺标样,结果如图2。从图2可以看出,氨基酸电位波形时,0.2 mL/min柱后补碱NaOH溶液的响应值高于0.3 mL/min柱后补碱NaOH溶液。若柱后补液流速到0.1ml/min,由于淋洗液为酸,补液为强碱,过低的补液流速和淋洗液的混合效果不好,且流量精度会降低,导致噪音变大。因此实验选择柱后补碱NaOH溶液流速为0.2 mL/min。[align=center][img=,690,531]https://ng1.17img.cn/bbsfiles/images/2019/08/201908312150354680_6766_3426139_3.jpg!w690x531.jpg[/img][/align][align=center][b]图2 柱后补液流速的影响[/b][/align][align=center]Fig.2 Effect of post-column rehydration flow rate[/align][b]2.2 方法学验证[/b]2.2.1 线性关系、检出限和定量限本实验考察0.132 2-1.322 mg/L 范围内三乙胺的线性关系。待仪器稳定后,将配制的标准系列溶液由低浓度到高浓度顺序依次进样,平行测定三次,计算峰面积并取平均值。结果表明,三乙胺的线性关系良好,回归方程为y=1.076x+0.344 5,R2为0.999 4。三乙胺检测方法的检出限浓度为0.045 mg/L,相当于样品检出限含量为18.2 mg/kg,定量限浓度为0.15 mg/L,相当于样品的定量限含量为60.7 mg/kg。2.2.2 标准溶液进样重复性取三乙胺测定线性关系中浓度为0.661 0 mg/L的标准溶液作为进样重复性溶液,连续测定6次,记录峰面积。结果显示测得三乙胺峰面积的RSD为1.9 %(n=6),说明该分析方法较稳定,具有较好的进样重复性。2.2.3 实际样品分析取三批供试品,配制好实际样品溶液(约2500 mg/L),按上述色谱条件,对实际阿立哌唑样品进行检测,色谱图见图3。从图3中可以看出,阿立哌唑供试品中未检测到三乙胺毒性杂质,小于18.2 mg/kg。[align=center][img=,690,478]https://ng1.17img.cn/bbsfiles/images/2019/08/201908312150529972_3514_3426139_3.jpg!w690x478.jpg[/img][/align][align=center][b]图3 实际样品色谱图[/b][/align][align=center]Fig.3 Chromatogram of the actual sample[/align]2.2.4 加标回收实验对供试品1[sup]#[/sup]中成分三乙胺进行回收率实验。精密量取2.5 mL浓度为2500 mg/L的实际样品溶液分别置于5 mL容量瓶中,分别精密加入2.5 mL浓度为1.322 mg/L、0.661 0 mg/L、0.440 7 mg/L的对照品储备液,混合均匀。在上述色谱条件下进样测定,每个浓度平行测定三次,回收率结果见表1。从表1中可以看出,样品不同水平加标回收率在101.7%~105.9%之间,说明该检测方法可信度较高。[align=center][b]表1 样品加标回收率[/b][/align][align=center]Table 1 Results ofrecovery tests for sample[/align] [table=657][tr][td] [align=center]化合物[/align] [align=center](compound)[/align] [/td][td] [align=center]样品含量[/align] [align=center](sample amount)/( mg/L)[/align] [/td][td] [align=center]加标量[/align] [align=center](addition)/( mg/L)[/align] [/td][td] [align=center]检测量[/align] [align=center](measured amount)/( mg/L)[/align] [/td][td] [align=center]回收率[/align] [align=center](recovery)/%[/align] [/td][/tr][tr][td=1,3] [align=center]三乙胺(Triethylamine)[/align] [/td][td] [align=center]-[/align] [/td][td] [align=center]0.066 1[/align] [/td][td] [align=center]0.070 0[/align] [/td][td] [align=center]105.9[/align] [/td][/tr][tr][td] [align=center]-[/align] [/td][td] [align=center]0.132 2[/align] [/td][td] [align=center]0.134 4[/align] [/td][td] [align=center]101.7[/align] [/td][/tr][tr][td] [align=center]-[/align] [/td][td] [align=center]0.220 4[/align] [/td][td] [align=center]0.232 4[/align] [/td][td] [align=center]105.4[/align] [/td][/tr][/table][align=center][/align][b]3 结论[/b]上述实验结果表明,采用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱后补液-安培法,在30 mmol/L的甲烷磺酸溶液淋洗液,流速为1 mL/min,柱后补碱为500 mmol/L的NaOH溶液,流速为0.2 mL/min,氨基酸电位波形的色谱条件下,能准确地分析阿立哌唑中基因毒性杂质三乙胺的残留量,最低检出限为18.2 mg/kg,灵敏度高,满足药典的要求,该方法准确度、精密度和稳定性较好。在阿立哌唑的质量控制中,该方法对三乙胺残留量的控制有重大意义。[b]参考文献:[/b] Zhang P, Li Z D, Jiao Z. Second generation atypical antipsychoticdrug aripiprazole. Chin Pharm J, 2005, 40(3): 238-240.张璞,李中东,焦正.第二代非典型抗精神病药一阿立哌唑. 中国新药杂志,2005,40(3):238-240. Wu C Y, Zhu Y C. Synthesis ofaripiprazole. Chin J Mod Drug Appl, 2010, 4(1): 11-12.吴春艳,朱永超. 阿立哌唑的合成 . 中国现代药物应用,2010,4(1):11-12. Li M D, Cai J, JiM. Synthesis of atypical antipsychotic new drug aripiprazole. Prog PharmSci, 2004, 28(6): 274-276.李铭东,蔡进,吉明. 非典型抗精神病新药阿立哌唑的合成.药学进展,2004,28(6):274-276. Liu X J, Wang T T,Zhong Y L, et al. Synthesis of antipsychotic aripiprazole. JShengyang Pharm Univ, 2013, 30(4): 253-255.刘秀杰,王媞媞,钟永亮,等. 抗精神病新药阿立哌唑的合成. 沈阳药科大学学报,2013,30(4):253-255. Xu J M, Wu Q Y,Zhang J, et al. Research on preparation of aripiprazole. J PharmPractice徐建明,吴秋业,张俊,等. 阿立哌唑的制备工艺研究. 药物实践杂志,2005,23(5):269-270. Ge H X, Wang L C,Ni S L. Improved Synthesis of Antipsychotic DrugAripiprazole. Chin JMAP, 2007, 24(4): 294-295.葛海霞,王礼琛,倪生良. 抗精神病药阿立哌唑的合成工艺改进. 中国现代应用药学,2007,24(4):294-295. Chen G Y, Chen X B,Liu G M, et al. Improved Synthesis of Aripiprazole. Shandong Chem Ind,2009, 38(9): 3-5.陈光勇,陈旭冰,刘光明,等. 阿立哌唑的合成工艺改进. 山东化工,2009,38(9):3-5. ICH guideline Q3C (R6 on impurities: guideline for residual solvents. (2016-12). Ma Z Q, Lu G H, YinJ J, et al. Determination of Residual Triethylamine in Rupatadine Fumarate byGas Chromatography. Guangdong Chem, 2018, 45(14): 213-214.马振千,鹿贵花,印嘉佳,等. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定富马酸卢帕他定中三乙胺的含量. 广东化工,2018,45(14):213-214. Guo J T, Yu F.Determination of Triethylamine Residue in Tetraethyl Ammonium Bromide AqueousSolution by Capillary Gas Chromatography. Guangdong Chem, 2018, 45(14):213-214.郭建亭,于飞. 毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定四乙基溴化铵水溶液中三乙胺的残留. 化学世界,2017,12:727-729. Wu W P, Ming D.Determination of Triethylamine Residues in Raw Material of Mirtazapine by GasChromatography. Yunnan Chem Tech, 2018, 45(8): 130-131.伍蔚萍,明丹. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定米氮平原料药中三乙胺残留量. 云南化工,2018,45(8):130-131. Jin Z Q.Discussion on problems of water Triethylamine determination. Tech Wind,2014, 23: 108.金梓谦. 分光光度法测定水中三乙胺有关问题探讨. 科技风,2014,23:108. Chen Z, Wang D,Zhang J N, et al. Optimization of Detection Technology for TriethylamineHydrochloride in Industrial Effluent. Salt Sci and Chem Ind J, 2019, 48(3):29-32.陈峥,王丹,张金娜,等. 工业废水中三乙胺盐酸盐检测技术的优化. 盐科学与化工,2019,48(3):29-32. Chen M S, Liang Z,Tang H Y, et al. Simultaneous determination of the migration of five alcoholamines in plastics food contact materials and articles by non-suppressed ion chromatography.AL, 2018, 37(10): 1183-1188.陈旻实,梁震,唐寰宇,等. 非抑制[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定塑料食品接触材料中五种醇胺迁移量. 分析试验室,2018,37(10):1183-1188. Fang L M, Hu M,Chen A L, et al. Determination of the residual dimethylamine in arbidolhydrochloride by ion chromatography. Chin J Pharm Anal, 2016,36(10):1852-1856.方琳美,胡咪,陈爱连,等. [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定盐酸阿比朵尔中残留的二甲胺含量. 药物分析杂志,2016,36(10):1852-1856. Li X C, Ding M M,Cai Q, et al. Determination of Triethylamine in Glyphosate Synthesis by IonChromatography. Qingdao: Proceedings of the 13th Ion Chromatography AcademicConference. 2010.李向春,丁敏敏,蔡琪,等. [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定草甘膦合成工艺中的三乙胺. 青岛:第13届[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]学术报告会论文集. 2010. Pan S, Shi C O,Liu Y M, et al. Determination of hydroxylamine hydrochloride in micafunginsodium by ion chromatography with pules amperometric detection. Anal Instru,2018(2): 58-61.潘思,施超欧,刘玉梅,等. [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱后补液-积分脉冲安培法检测米卡芬净钠中残留盐酸羟胺. 分析仪器,2018(2):58-61.
当前开展丙二醛 2,4-二硝基苯肼柱前衍生液相色谱310nm检测,流动相0.2%乙酸-乙腈(1:1),结果只有 2,4-二硝基苯肼峰,未见明显衍生产物峰,没有衍生好是什么原因。衍生方法为丙二醛在高氯酸或三氯乙酸酸性条件衍生。
我用DB-624柱子FID检测器测定三乙胺,峰总是严重拖尾,请问各位大侠怎么解决?非常着急,谢谢!
如题,检测室最近购买了一台安捷伦1260液相色谱仪,配的是荧光检测器和二极管阵列检测器,钱不够,就没安柱后衍生系统,我们是做农产品及水质类的农药残留检测的 ,查了下资料,像安基甲酸酯类的农药,基本都要求用紫外检测器,我想请教各位高手,像我们现在这样的配置,可以测点啥呢。谢谢了。
最近在用异硫氰酸苯酯(PITC)检测氨基酸的时候出现了一个怪问题:17个氨基酸只有前面9种氨基酸可以出峰,剩下的氨基酸都不能出峰。液相条件什么都是一样的,换了新柱子问题仍然存在。一开始怀疑由于现在天气凉了衍生不充分,就放在25℃水浴中衍生2小时,问题仍然存在。让我很是苦恼。调整梯度程序、温度都不管用。液相也很正常。下面是我的液相条件:请各位高手帮忙啊!5色谱条件色谱柱: Sepax AA专用柱,4.6*250mm,由苏州赛分科技有限公司提供。检测波长:254nm。柱温:40℃。流动相A: 醋酸钠溶液:乙腈=93:7 (V/V)。流动相B: 乙腈:水=4:1(V/V)。进样量:5ul流速:1mL/min(注:分析结束后使用20%乙腈水溶液清洗,再用100%乙腈清洗后储存) 表1 梯度洗脱程序 时间/min 流动相A/% 流动相B/% 0 100 0 2 100 0 14 93 7 29 70 30 32 50 50 33 0 100 39 0 100 39.1 100 0 45 100 0 6 氨基酸标准溶液和样品溶液的衍生(设2个重复)6.1 17AA衍生准确量取氨基酸标准溶液400μl,置5.0ml塑料离心管中,加入三乙胺乙腈溶液(2.10)200μl,异硫氰酸苯酯乙腈溶液(2.11)200μl,混匀,室温放置1小时,然后加入正己烷800μl,振摇后放置10分钟,取下层溶液(PTC-AA),用一次性注射器吸取上清液用0.45μm针式过滤器过滤。准确量取样品溶液400μl,置5.0ml塑料离心管中,加入三乙胺乙腈溶液(2.10)200μl,异硫氰酸苯酯乙腈溶液(2.11)200μl,混匀,室温放置1小时,然后加入正己烷800μl,振摇后放置10分钟,取下层溶液(PTC-AA),用一次性注
安捷伦正相液相90毫升正己烷和10毫升异丙醇加0.1毫升二乙胺,用双通道的,这0.1毫升二乙胺都加在了异丙醇中,基线会漂移,怎么办?0.1毫升二乙胺到底怎么加
[size=4][font=KaiTi_GB2312] 游离棉酚是棉籽饼中含有的一种有毒物质,可以杀灭精子,用于男性避孕。因棉籽饼含大量蛋白质,常用作动物饲料,但其所含的棉酚对动物有抑制生长的作用。检测棉酚含量的标准方法一般为紫外分光光度检测和高效液相色谱法,目前也有用LC/MS/MS法进行检测的报道,但尚未见使用GC/MS法检测的报道。 因棉酚挥发性和稳定性较差,分子结构为含双萘环的多羟基化合物,所以我尝试使用衍生化进行检测。首先使用了硅烷化试剂进行衍生,结果出现多个色谱峰,谱图如下:[img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003061118_204192_1608554_3.jpg[/img] 从图中可以看出在7.48、7.55和11.96min出现了三个峰,峰的质谱图如下:[img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003061121_204193_1608554_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003061121_204194_1608554_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003061123_204196_1608554_3.jpg[/img]第三个峰的谱库检索显示为邻苯二甲酸酯。 其后我又使用乙酰化衍生,色谱图如下:[img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003061125_204197_1608554_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003061125_204198_1608554_3.jpg[/img]11.95min的质谱检索显示仍为邻苯二甲酸酯。 而棉酚的分子量为578.6,我非常困惑,这是为什么?这个峰是污染的吗?可是我用纯溶剂并不能跑出这个峰啊![/font][/size]
求助!最近在做胺类化合物的检测,用的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url],色谱柱是kbs-wax,进样量1ml,柱温70℃,进样口120℃,检测器180℃,最开始出峰都挺好的,就是后面副产物有点分不开,但是慢慢进口和标气单一物质打进色谱峰形变得很差,最后直接不出峰,(图1-图2的变化)就很小很小几百的峰面积。想问大家在做胺类物质的时候出峰也会这样变差吗?如果想检测正丁胺和二乙胺这两种物质,最好用什么色谱柱呢?[img]https://ng1.17img.cn/bbsfiles/images/2022/07/202207092033288835_3538_5550972_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/07/202207092033299812_7633_5550972_3.png[/img]
RT,请教国标气质法检测三聚氰胺,衍生后生成什么?
苯乙胺采用最新国标衍生方法衍生后(只是没有用乙醚萃取),采用荧光光度计测量荧光值,值十分小,为什么?
想请教一下,如果采用衍生气象色谱内标法分析时,内标物如何选择呢?是在优化衍生条件时就加入内标呢?还是在优化衍生条件之后,作方法的分析性能的时候再加入内标呢?因为我的内标必须同时经过衍生才能进入气象色谱分析,内标本身没有挥发性,需要衍生,才适合GC分析。我是新手,没做过衍生也不知道过量的衍生试剂如何除去?请各位帮帮忙!
按文献方法检测发现样品图谱中有三个峰,无法确定哪个是对照品峰,衍生试剂为2,4-二硝基苯肼?目的是为了检测饲料用油脂中丙二醛含量,国标法用的是硫代巴比妥酸衍生,但用的是荧光检测器当前手上没有,只有紫外检测器







 我要推广仪器
我要推广仪器
 下载APP
下载APP