福建13家医疗机构制剂室检查 仪器成了“重灾区”
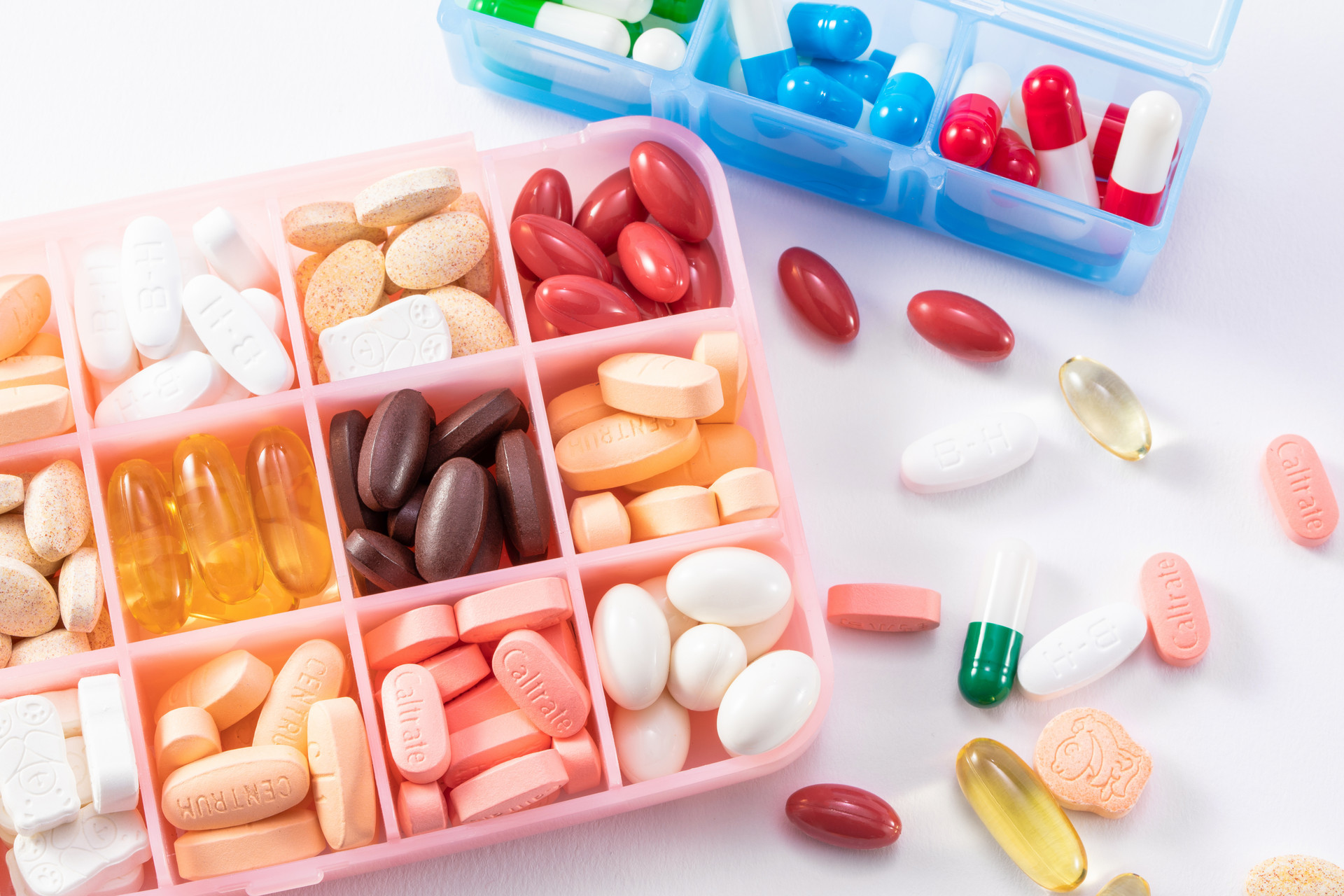
p span style=" font-family: 楷体,楷体_GB2312,SimKai " 日前,按照福建省食品药品监督管理局开展医疗机构制剂室专项整治的工作部署及《医疗机构制剂配制质量管理规范(试行)》的要求,福建省食品药品监督管理局按双随机方案组织5个检查组分赴厦门大学附属第一医院等13家医疗机构,就制剂室人员、设施设备、物料、卫生、配制管理、质量管理等管理情况进行了专项整治检查。 /span /p p span style=" font-family: 楷体,楷体_GB2312,SimKai " 本次检查共发现缺陷139项,相关需要说明问题6项,其中约10项涉及分析仪器的使用不当或不具备。福建省食药监局此次发布的检查结果中,分析仪器也成了“重灾区”: /span /p p strong 1、药检室中电导率仪、安捷伦7890AGC无仪器状态标识,部分仪器缺合格标志。 /strong /p p strong 2、依沙吖啶溶液(批号170519)含量测定中,依沙吖啶乳酸盐对照品未恒重,未使用相适应的电子天平 紫外分光光度法缺原始图谱 药检室未按规范使用预检定的滴定管 部分制剂未按规范进行微生物限度检测。 /strong /p p strong 3、药检室仪器使用记录不完整,如pH计使用记录中,无样品的批号。 /strong /p p strong 4、化验室未配置纯化水检验用所需电导率检测仪器。 /strong /p p strong 5、未制定纯化水制备系统使用、维护、保养管理制度,未定期更换纯水机滤芯。 /strong /p p strong 6、部分仪器不符合物料检验要求,如复方薄荷脑滴鼻液成品的樟脑鉴别所用的紫外分光光度计(型号7520)不能在230nm到350nm波长间进行扫描,不能精确测定吸收值峰值。用于呋麻滴鼻液成品的盐酸麻黄碱含量测定所使用的旋光仪(型号WZM001)不能正常使用。 /strong /p p br/ /p p strong 7、纯化水系统制水设备无状态标识。 /strong /p p strong 8、缺部分物料检验所需要仪器设备,如用于复方呋喃西林滴鼻液成品的盐酸麻黄碱含量测定的旋光仪,用于隆力福胶囊成品的三七皂苷R1含量测定的高效液相色谱仪。 /strong /p p strong 9、用于中药饮片粉碎的设备无设备铭牌,无状态标识。 /strong /p p strong 10、药检室未配备电导率仪。 /strong /p p span style=" color: rgb(255, 0, 0) " strong 附缺陷汇总清单: /strong /span /p p span style=" color: rgb(0, 112, 192) " strong 一、现场检查厦门大学附属第一医院发现缺陷项目10项: /strong /span /p p 1、该制剂室配制的品种及批次较多,制剂室与药检室专业技术人员偏少。 /p p 2、制剂室部分人员接受本规范培训效果欠佳。 /p p 3、固体制剂车间面积偏小。 /p p 4、液体制剂分装间台面与墙壁之间有裂缝 制材制粒粉碎间墙体上有3个普通插座 固体制剂车间烘干间有一台家用分体式空调。5、洁净区内配制内服与外用的纯化水软管未严格区分。 /p p 5、洁净区内配制内服与外用的纯化水软管未严格区分。 /p p strong 6、药检室中电导率仪、安捷伦7890AGC无仪器状态标识,部分仪器缺合格标志。 /strong /p p 7、成品库中,各成品缺明显的状态标识 电脑管理的台账未按批号进行区分。 /p p 8、配制间洁净室及药检洁净室内清洁不同部位的毛巾未进行严格区分 清洁用刷子易产生脱落物 洁净区内已清洁的配液罐及搅拌罐内部有水珠 样品实验室高效过滤器表面大量粉尘。 /p p 9、甘安合剂与呋麻滴鼻液的批配制记录中,称量及投料工序缺对特殊药品原料(复方樟脑酊、盐酸麻黄碱)监控记录 肚液散(批号20161215)批配制记录中,粉碎工序与混合工序缺使用的设备名称、型号、编号的记录。部分文件制定前未进行认真核对,部分文字内容有误,如:《氧化锌Ⅱ号洗剂工艺规程》(编号:PR-TS-01-052-01)中,氧化锌处方量有误 《3%薄荷乳膏工艺规程》(编号:PR-TS-01-057-01)所写的处方用量与批准的处方用量不同。 /p p 10、成品检验原始记录存在较多缺陷,如:3%薄荷乳膏(批号170506)薄层鉴别缺原始图谱,装量原始记录不完整,数据不原始 strong 依沙吖啶溶液(批号170519)含量测定中,依沙吖啶乳酸盐对照品未恒重,未使用相适应的电子天平 紫外分光光度法缺原始图谱 药检室未按规范使用预检定的滴定管 部分制剂未按规范进行微生物限度检测。 /strong /p p span style=" color: rgb(0, 112, 192) " strong 二、现场检查福建省漳州市皮肤病防治院发现缺陷项目11项: /strong /span /p p 1、制剂配制人员、药检人员接受本规范培训效果不佳。 /p p 2、洁净区洗手池的下水管封闭不严。 /p p 3、洁净区内的温湿度计、压力表、压差计无检定的标识 玻璃器皿自检记录不完整。 /p p strong 4、药检室仪器使用记录不完整,如pH计使用记录中,无样品的批号。 /strong /p p 5、标签发放记录中,发放人员未签名。 /p p 6、固体物料与液体物料未合理分区存放,可能造成交叉污染。 /p p 7、已清洁的不锈钢配制桶内有少量残留水。 /p p 8、已消毒的洁净服没有包装,没有标明有效期。 /p p 9、文件缺生效及执行日期 已过时的文件,如:蒸馏水器使用操作规程(编码:SOP-M015)未及时销毁 文件未及时更新,多数技术标准中,工艺用水仍写为“蒸馏水” 2015年度配制记录和检验记录未按规定保存2年备查。 /p p 10、制剂规程中无不合格品的处置程序及监督措施。 /p p 11、制剂室规章制度中,半成品的内控标准及检验操作规程不完整 无培养基管理方法 洁净区的微生物数未按规章制度进行检测。检验原始记录存在缺陷,如:炉甘石硫洗剂(批号:20170601)装量检测错误 硼酸溶液(批号:20170601)含量测定计算错误 复方氯霉素洗剂(批号:20170601)pH值测定错误 曲安缩松乳膏(批号:20170501)装量检测取样错误。 /p p span style=" color: rgb(0, 112, 192) " strong 三、现场检查泉州市第一医院发现缺陷项目11项: /strong /span /p p 1、原辅料库、包材库面积较小,不能分类、分区、离墙存放,较拥挤。 /p p 2、2016年度《空气洁净度检测报告书》(报告编号:201645)部分项目不符合规范要求,如内服配制间、内服分装间换气次数不符合规范要求(只达12次) 化糖间0.5微米的悬浮粒子超标 主要操作间的照度均达不到300Lx,未采取相应的整改措施。 /p p 3、外墙进入化糖间的蒸气管道与墙壁连接部位未密封。 /p p 4、微生物限度检查缺少相应的阳性对照间。 /p p 5、物料无入库验收记录、物料货位卡,部分物料领用记录不规范,物料无法按批号进行追溯。 /p p 6、复方樟脑酊、颠茄酊未按要求阴凉储存。 /p p 7、内服器具洗涤间使用铁丝球做为器具洗涤工具。 /p p 8、配制规程和配制记录中缺少具体的配制参数,如单糖浆配备过程中缺少加热温度、加热时间等参数。 /p p 9、未制订原辅料和内包材的检验操作规程 成品检验记录缺性状和装量检查项目。 /p p 10、部分管理文件从2006年至今未审核修订。 /p p 11、滴定液未按要求进行双人标定。 /p p span style=" color: rgb(0, 112, 192) " strong 四、现场检查泉州市正骨医院发现缺陷项目15项: /strong /span /p p 1、部分人员按《医疗机构制剂配制质量管理规范》培训不到位。2、洁净区内缺少相应的酒剂、搽剂的灌装间,器具和洁具洗涤共用一个功能间 药材净制、炼蜜、炸枯、炼油工序在同一操作间操作 中药饮片烘干使用的HX-20热风循环烘箱位于附属楼五层楼梯口 缺少一般区的更衣区域。 /p p 3、中药饮片和制剂成品存放于同一库房 内包材库通风防潮措施不足,现场检查时发现有霉味 /p p 4、洁净区内的干燥间和清洁间内表面不光滑。 /p p 5、洁净区内空气洁净度监测未按《洁净区使用、管理和监测制度》执行。2015年度《空气洁净度检测报告书》部分项目不符合规范要求,如总混间、制丸间一、制丸间二、缓冲间换气次数不符合规范要求,主操作间照度达不到300Lx。 /p p 6、制水车间位于洁净区内,制水车间与提取车间之间的墙壁存在多处的缝隙。 /p p 7、洁净区与一般区的压差达不到10帕。 /p p 8、空调净化系统新风采集室内风,部分新风过滤后进入初效前段,部分新风与回风混合后直接进入中效前段,初效、中效两端未安装压差装置。 /p p 9、纯化水采取二级反渗透制备,但缺少一级反渗透水储罐。 /p p 10、微生物限度检查缺少相应的阳性对照间。 /p p 11、物料无入库验收记录、物料货位卡 伤科搽剂和正骨活络油未按阴凉条件储存 贮存于内包材中转间的空心胶囊无标识,从2016年12月2日使用完后一直放置于内包材中转间。 /p p 12、胶囊填充机清洁不彻底,有残留的空心胶囊 胶囊填充间高效口有残留药物粉尘。 /p p 13、《生产操作规程》中散剂品种的工艺流程与《福建省医院制剂配制规程》不一致,在低温间歇灭菌工序前增加了一道烘干工序。 /p p 14、竭七胶囊配制中三七净制后无称量记录。 /p p 15、该制剂室无相关的自检记录。 /p p span style=" color: rgb(0, 112, 192) " strong 五、现场检查福建医科大学孟超肝胆医院(福州市传染病医院)发现缺陷项目15项: /strong /span /p p 1、制水岗位操作人员操作技能培训效果不佳。 /p p 2、洁净室内个别功能间现场发现有蚊子 中药饮片仓库门缝较大,防止昆虫和其他动物进入措施不足。 /p p 3、内服器具洗涤存放间水池与地面接口出现开裂 洁具洗涤存放间地面有霉迹。 /p p 4、未配置鲜金线莲榨汁设备。 /p p 5、部分已停用设备未及时搬出生产场所,如:DTQ新型自控多功能中药提取器、多功能无级调程自动泡罩包装机等 中药提取车间用于提取液过滤的筛网已破损,未及时更换 中药提取车间的蒸汽输送管道部分保温材料脱落,裸露部位金属材料生锈腐蚀较为严重,存在安全隐患。 /p p 6、未建立物料台账 物料采用货位卡管理,但已使用完的物料货位卡未归档,无法对物料进行追溯。 /p p 7、未规定鲜金线莲及鲜金线莲汁、中药提取液的储存条件及储存期限。 /p p 8、降酶灵胶囊标签、说明书中功能与主治未按省局新颁布实施的医疗机构制剂规程相应内容及时更新。 /p p 9、现场未能提供洁净室地漏定期消毒记录,地漏液封出现混浊及颜色发黑的情况,个别地漏有蟑螂出没。 /p p 10、抗纤I号片配制操作规程制定内容不完整,未制定相关操作工序的技术参数,可操作性不强。) /p p 11、现场使用的《YG.10B易拉瓶自动灌装机操作规程》无制定、审查和批准人的签名。 /p p 12、磨粉设备清场不彻底,现场发现设备内部有残留物 YG 10B易拉瓶自动灌装机灌装部位(含玻璃活塞、硅胶管)清洗不彻底 100万CC热回流提取浓缩回收机组中的提取罐内的管道出口有霉迹。 /p p 13、批制剂记录内容不完整,个别配制环节未在批记录中体现。 /p p 14、部分制剂用的原辅料未按标准进行检验后使用。 /p p 15、自检记录未见评价及改进措施。 /p p span style=" color: rgb(0, 112, 192) " strong 六、现场检查福州市皮肤病防治院发现缺陷项目11项及相关需要说明问题4项: /strong /span /p p 1、仓库管理人员及检验人员对本岗位岗位技能培训效果不佳。 /p p 2、未单独设置成品库,现场检查时成品存放于外包装间 包材库面积偏小,不能满足生产实际需要。 /p p 3、酊剂分装间部分墙面有霉迹。 /p p strong 4、化验室未配置纯化水检验用所需电导率检测仪器。 /strong /p p 5、物料货位卡内容不完整,未体现规格、有效期等信息 货位卡未按产品批号分开填制,无法对物料去向进行追溯 包材仓库内用于洗剂、溶液剂内包装的塑料瓶未密封存放 个别物料在仓库内称量发放,如单、双硬脂酸甘油酯。 /p p 6、成品存放场所未设置温湿度调控及监测设备,需阴凉储存的薄荷脑、鱼肝油等原料存放场所温度超标(现场温湿度显示装置显示温度为27℃)。 /p p 7、复方硫磺洗剂2号标签中主要成分标识有误:把“硫磺”误写成“雷磺” 补骨脂酊标签中“注意事项”项下内容与注册批件不一致。 /p p 8、检查发现补骨脂酊、含酚炉甘石洗剂和尿素乳膏未制定中间品检验规程。 /p p 9、留样观察室工作制度无文件制定人、复核人及批准人签字及批准日期。 /p p 10、检查发现补骨脂酊、含酚炉甘石洗剂和尿素乳膏批检验记录“检查”项未检验 部分制剂用原辅料未按标准检验后使用。 /p p 11、2016年自检记录缺问题评价及改进措施。 /p p 需要说明问题4项: /p p 1、在省局已发布的“福建省医疗机构制剂规程1-8批品种目录公告”中,涉及该院制剂品种13个。该院未按照文件要求,对涉及的制剂品种进行相关文件(如:制剂规程、检验规程、标签说明书)的修改。 /p p 2、酊剂配制间内使用的抽滤装置未配置防爆型电机,存在安全隐患。 /p p 3、该院制剂成品及纯化水的微生物限度检验项目委托本院化验室检验。 /p p 4、该院部分制剂品种(地塞米松冰片乳膏、复方氢化可的松乳膏、氢松鱼石糊、粘膜溃疡脂、地塞米松磷酸钠乳膏)使用激素类原料配制,与其他制剂品种共用生产设备。 /p p span style=" color: rgb(0, 112, 192) " strong 七、现场检查三明市皮肤病医院发现缺陷项目13项及相关需要说明问题2项: /strong /span /p p 1、制水岗位操作人员及检验人员对本岗位技能培训效果不佳。 /p p 2、制剂室现有面积、洁净区功能间与所配制制剂剂型和规模不相适应 制剂室未配备产品阴凉留样场所。 /p p 3、制剂配制用95%乙醇存放场所排气扇不能启动,照明设施及其开关无防爆功能 原辅料常温库内设有一厕所,防污染措施不到位。 /p p 4、未对洁净室内空气的微生物数和尘粒数开展定期检测。 /p p 5、洁净区内灌装间与空调机房出入门加锁关闭,门下部有一回风口与外界直接相通。 /p p 6、制剂室现有纯化水制备设备产量不能满足现有制剂配制需要。 /p p strong 7、未制定纯化水制备系统使用、维护、保养管理制度,未定期更换纯水机滤芯。 /strong /p p 8、个别物料在仓库内称量发放,如硬脂酸。 /p p 9、地塞米松冰片乳膏、硼酸洗剂、炉甘石硫磺洗剂说明书的“适应症”、“用法用量”等内容与省局发布的规程不一致 所有制剂品种的标签、说明书无“本制剂仅限本医疗机构使用”字样。 /p p 10、中间品未制定检验操作规程。 /p p 11、药检室留样管理规程(文件编号:JYSOP-007-00)对产品留样时间、样品销毁方式制定不科学。 /p p 12、对照品、滴定液未制定管理办法 部分原料未按法定标准检验,只进行了部分检验。 /p p 13、2017年自检记录缺问题评价及改进措施。 /p p 需要说明问题2项: /p p 1、在省局已发布的“福建省医疗机构制剂规程1-8批品种目录公告”中,涉及该院制剂品种11个。该院未按照文件要求,对涉及的制剂品种进行相关文件(如:制剂规程、检验规程、标签说明书)的修改。 /p p 2、该院部分制剂品种(地塞米松冰片乳膏、克氯乳膏、曲安缩松乳膏、复方克林霉素搽剂、粘膜溃疡脂、地塞米松新霉素糊)使用激素类原料配制,与其他制剂品种共用生产设备。 /p p strong span style=" color: rgb(0, 112, 192) " 八、现场检查福建省南平市第一医院发现主要缺陷项目1项,一般缺陷9项: /span /strong /p p 主要缺陷为部分原辅料、半成品未开展取样及检验工作。 strong 部分仪器不符合物料检验要求,如复方薄荷脑滴鼻液成品的樟脑鉴别所用的紫外分光光度计(型号7520)不能在230nm到350nm波长间进行扫描,不能精确测定吸收值峰值。用于呋麻滴鼻液成品的盐酸麻黄碱含量测定所使用的旋光仪(型号WZM001)不能正常使用。 /strong /p p 一般缺陷为 /p p 1、纯化水管道上穿洁净室顶部处部分接口不严密,有缝隙 洁净走廊墙壁与地面交界处的部分弧形接口开裂。 /p p 2、空气净化系统维护保养不到位,无初中效清洗更换相关记录,空调净化机组间存放大量杂物。 /p p 3、不合格品区设置在楼道走廊,未上锁管理。 /p p 4、部分配制用设备、容器无清洁状态标识。 /p p 5、纯化水管道未标识内容物及流向。 /p p 6、部分成品(如水合氯醛)未按要求阴凉储存。 /p p 7、外用工具间存放的标准筛清洗不彻底,残留少量白色粉末。 /p p 8、50%硫酸镁溶液(批号20170207,批量40000ml)配制记录不完整,缺少分2次配制20000ml,再混合成40000ml的配制过程记录。 /p p 9、未制定液体定量灌装机(型号ZYG30ml)的清洁标准操作规程。 /p p span style=" color: rgb(0, 112, 192) " strong 九、现场检查南平市疾病预防控制中心皮肤病性病防治院中心门诊部发现主要缺陷项目1项,一般缺陷10项: /strong /span /p p 主要缺陷为: /p p 部分制剂品种未制定原辅料、半成品的质量标准和检验操作规范。部分原辅料、半成品未开展取样及检验工作。 /p p 一般缺陷为: /p p strong 1、纯化水系统制水设备无状态标识。 /strong /p p 2、空气净化系统维护保养不到位,男一更间安装的压差计损坏未及时更换。 /p p 3、、物料仓库存放的物料无状态标识,未划分不合格品区。 /p p 4、原辅料库和成品库无相关温湿度记录,无降温除湿设备和防虫纱窗,成品库无货位卡并存放无关的杂物。 /p p 5、部分配制用设备、容器无清洁状态标识。 /p p 6、配制规程内容不够完整,缺部分具体操作步骤和技术参数等内容。配制记录设计内容不完整,配制过程的记录内容不全。 /p p 7、检验记录书写不规范,如有效数字使用不规范,含量检测项目未计算相对标准偏差。成品硼酸氧化锌粉(批号170508)检验记录缺干燥失重项目检测的数据。 /p p 8、洁具清洗间地漏无消毒液液封,用水管道未标识内容物及流向。 /p p 9、部分成品和原辅料(如氯倍他索尿素软膏、鱼肝油)未按要求阴凉储存。 /p p 10、FZ-1型半自动乳膏定量灌装机未制定标准清洁操作规程。 /p p span style=" color: rgb(0, 112, 192) " strong 十、现场检查福建省南平市人民医院发现主要缺陷项目1项,一般缺陷8项: /strong /span /p p 主要缺陷为: /p p 部分用于制剂的中药材(如三七)、中药饮片(如当归、炙甘草)未开展取样及检验工作。部分制剂品种(如大黄微粉胶囊)缺直接入药的中药粉末入药前的微生物限度检查。 strong 缺部分物料检验所需要仪器设备,如用于复方呋喃西林滴鼻液成品的盐酸麻黄碱含量测定的旋光仪,用于隆力福胶囊成品的三七皂苷R1含量测定的高效液相色谱仪。 /strong /p p 一般缺陷为: /p p 1、口服液灌装间墙壁与地面交界处的部分弧形接口开裂 蒸汽管道上穿洁净室顶部处接口不严密,有缝隙 更衣间、收膏间的洗手池表面锈蚀。 /p p 2、空气净化系统维护保养不到位,无初中效清洗更换相关记录,空调净化机组间存放大量杂物,男更衣室、女更衣室安装的压差计损坏未及时更换。 /p p 3、物料仓库存放的物料无货位卡,未划分不合格品区。 /p p 4、中药材三七外包装无产地、批号、采收日期等信息。 /p p strong 5、用于中药饮片粉碎的设备无设备铭牌,无状态标识。 /strong /p p 6、调配液罐(型号PYG-200)清洁不彻底,罐内残留少量的液体。 /p p 7、九九降压1号胶囊(批号20170606)批记录未记录中药饮片白芍、钩藤粉碎过筛和胶囊填充的过程。 /p p 8、中药材及中药饮片洗涤、浸润、提取用水未定期检验。 /p p span style=" color: rgb(0, 112, 192) " strong 十一、现场检查福建中医药大学附属人民医院发现主要缺陷项目2项,一般缺陷6项: /strong /span /p p 主要缺陷为 /p p 1、制剂室储存物料的库房面积与所配制制剂不匹配。购进的五桶口服液玻璃瓶盖存放于制剂大楼三楼走廊,未严格按照物料购进验收管理规程进行验收,产品外包装无厂家、批号等信息,且无货位卡 合格物料、待验物料及不合格物料未分别存放,且无明显标识,制剂大楼二楼原辅料暂存间内水杨酸等原料货位卡未记录批号 制剂大楼二楼洁净走廊内存放有12筐250ml玻璃瓶及两筐空试剂瓶 标签说明书存放于制剂大楼四楼空调机房内,未按品种、规格专柜(库)存放 /p p 2、药检室未能履行其主要职责: /p p (1)成品检验微生物限度检查项目未能进行阳性对照试验 /p p (2)未定期监测洁净室的微生物数和尘粒数 /p p (3)毒性试剂未建立购进使用台账 /p p (4)冰箱内存放已配制的胰酪大豆胨液体培养基、胰酪大豆胨琼脂培养基等未标注有效期。 /p p 一般缺陷为: /p p 1、菌检室空调系统关闭与打开两种状态下,一更与二更压差计指数均为10Pa /p p strong 2、药检室未配备电导率仪 /strong /p p 3、容器具存放间内已清洁容器无清洁状态标识 /p p 4、中药制剂配制规程未及时修订,批准日期为2005年,中药材质量标准仍为《中国药典》2005年版 化药制剂配制规程缺少原辅料、中间产品的质量标准、技术参数等,且无文件制定、批准人签名 /p p 5、《紫草油配制规程》(PZ-Sop-65-00)规程规定加热前浸渍半天,紫草油(批号:170325)配制记录浸渍时间为48小时,且未记录浸渍起始、结束时间 /p p 6、主要原辅料来源发生变更时未进行再验证。 /p p span style=" color: rgb(0, 112, 192) " strong 十二、现场检查福建医科大学附属第一医院发现主要缺陷项目1项,一般缺陷7项: /strong /span /p p 主要缺陷为: /p p 1、该院制剂微生物限度检查项目在该院检验科内完成,未对每批制剂及纯化水储罐用水进行微生物限度检验,无法提供样品交接记录,制剂微生物限度检验记录中检验数据的填写及检验人员签名均为制剂室工作人员 需进行微生物限度检查的成品未及时送检,如:复方氯霉素洗剂(批号:20161114)生产日期为2016年11月,微生物限度检验记录中检验日期为2017年4月7日,药品检验合格报告日期为2016年11月14日,制剂于2016年11月放行,但未记录制剂质量审核日期。 /p p 一般缺陷为: /p p 1、部分容器具存放于外用原料暂存称量间 /p p 2、内服原料称量间标准砝码已生锈未更换 /p p 3、未根据洁净式空调洗消规程(文件编号:GC-PZ-WS-KT)对空调系统进行清洁保养 /p p 4、原辅料货位卡未记录批号 /p p 5、留样间无温湿度调控设备 /p p 6、配制规程缺少原辅料、中间产品的质量标准、技术参数、包装材料的要求等信息 配制记录内容不全,如:克氯乳膏(批号:170613)未记录有关设备名称与操作记录、称量过程、温度控制参数等,装量差异检查未记录数据 /p p 7、外用制剂1功能间内配液罐(MF-100)清洁不彻底,罐体仍有白色粉末残留 /p p span style=" color: rgb(0, 112, 192) " strong 十三、现场检查宁德市中医院发现一般缺陷7项: /strong /span /p p 1、提取车间无防虫措施 /p p 2、工作间未按制剂工序要求合理布局,制粒工序与干燥工序共用功能间,摇摆式颗粒机与热风循环风箱共同放置在干燥间 菌检室与阳性对照检查室未分离,阳性间无传递窗 /p p 3、药剂科中药材仓库(制剂室所用中药材领用处)内荷叶(批号:1608099,浙江华宇药业股份有限公司)直接放置于地面 原药材及中药饮片未严格分区存放,已合格的中药饮片仍存放于待验区 未对原辅料库内存放的甘草流浸膏(批号:160923,福州海王金象中药制药有限公司)退库的数量进行复核 原辅料库内抽检后剩余的250g氧化锌粉(批号:20170101)包装袋敞口存放 水杨酸甲酯货位卡名称未使用药品通用名 /p p 4、配制规程不完善,未体现原辅料、包装材料的质量标准,个别工艺参数未制定,如脉康宁胶囊配制规程(SOP-PZ018-04-01)未规定提取液与粉末混合时间 配制记录内容不完整,如清淤排石颗粒(批号:170404)未记录烘干时间、烘干铺盘厚度、在线装量差异监测数据、熔封温度等,脉康宁胶囊(批号:170102)未记录混合起止时间 /p p 5、椎消突胶囊配制规程(SOP-PZ019-04-01)规定煎煮分为两次,每次1小时,与《福建省食品药品监督管理局关于发布福建省医疗机构制剂规程第三批品种目录的通告》中规定的煎煮分为两次,每次1.5小时不符 /p p 6、锌氧油(批号:170302)配制记录描述为在研钵中研匀,实际操作为在不锈钢盆中搅匀 /p p 7、玫瑰红钠琼脂培养基已过有效期未及时清除 滴定液标定记录未体现复标人操作。 /p p 福建省食品药品监督管理局要求辖区市食品药品监管(市场监督)部门督促相关医疗机构限期整改,同时应继续加强对医疗机构制剂的监督检查。 /p

























 我要推广仪器
我要推广仪器
 下载APP
下载APP