标准加入法对于ICP-MS是必须的,对于ICP-OES呢?大家的ICP-OES分析软件中有标准加入法这一程序?
大家购买水中有机物标准品是从哪些单位或者部门购买啊最近要购买一些水中 地下水有机物的标准品但是有些标准物质网买不到请问大家一般通过哪些渠道购买呢
有人分析过食品中有机磷么?是参照什么标准做的
标准曲线软件请教?
标准物质在分析实验室管理中有那些应用?一起谈谈。
全套锌化学分析标准,谢谢大家http://www.instrument.com.cn/download/shtml/054874.shtml
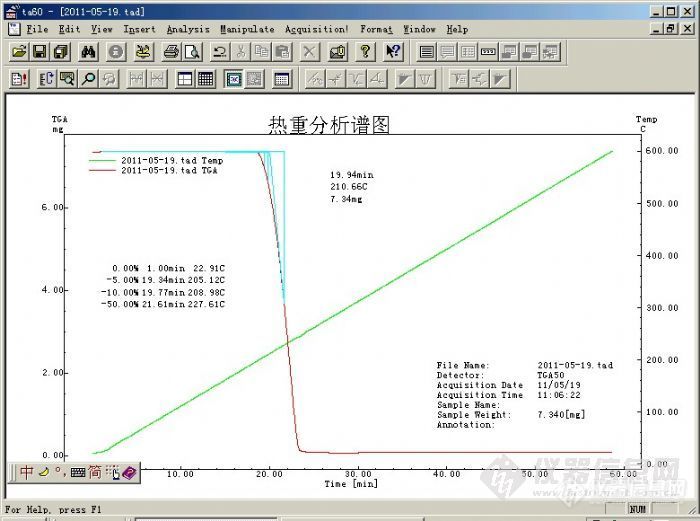
我没有看见说明书中有"平滑"设置最佳数值的建议,也没有相关资料,希望各位提供一些有关的参数和建议!我经过改变不同的"平滑"数值给一些是比较极端的大数字][/back]就是想看看"平滑"参数对分析结果的影响,数值达到一定时,谱图的形态和分析结果均有影响!!并且使用软件中的空白基线扣除功能数值大到起始基线或结束基线位置以上时谱图和分析数据影响较大,附上图片请大家给点建议1、热重分析谱图是否需要美化?2、基线是否需要扣除?扣除基线有何意义?[img]http://ng1.17img.cn/bbsfiles/images/2011/05/201105191819_294984_1750068_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2011/05/201105191818_294983_1750068_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2011/05/201105191819_294985_1750068_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2011/05/201105191820_294986_1750068_3.jpg[/img]原始的谱图是下一张[img]http://ng1.17img.cn/bbsfiles/images/2011/05/201105191821_294987_1750068_3.jpg[/img][/size]
大家使用的热分析的软件是中文的还是英文的呢?英文的是随机带的还是自己花钱买的呢?
大家在做响应面分析的时候用的什么软件呀?design-expert?还是其他的软件?哪个更好用呀?
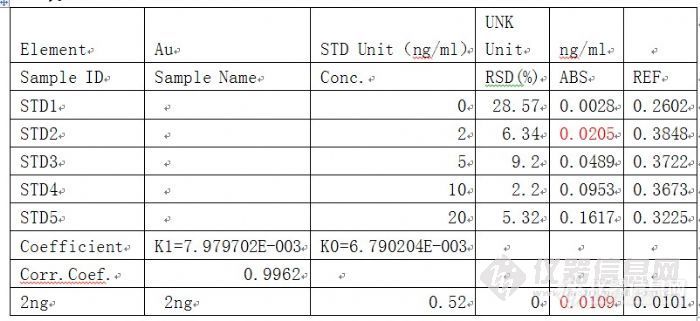
精密度不好。精密度长期不好(表1),有时调整针后,曲线做好了,RSD也不错,但是测样一段时间后,再测标准,发现吸光度值为原来的一半左右(表2)表1http://ng1.17img.cn/bbsfiles/images/2011/08/201108241738_312013_1601823_3.jpg 红色的RSD值太大。表2http://ng1.17img.cn/bbsfiles/images/2011/08/201108241741_312017_1601823_3.jpg注意红色标记,同一个标准,后来测的为原来的一半。请大家分析原因,主要有哪些。(石墨炉分析方法)
请问大家你们在用标准曲线法做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]或液相分析时多久重测标准曲线?
标准曲线法是仪器分析中最常用的方法,从标准来看GB/T22554-2010《基于标准样品的线性校准》中:1、标准曲线的浓度范围应覆盖正常操作条件下的被测量范围;2、标准样品的组分尽量与被测样品组分一致;3、标准样品的浓度值应等距离的分布在被测量范围;4、标准样品的个数至少应有3个浓度;5、每个标准点至少重复2次,这个重复是指从稀释开始;如果国家标准有相应的浓度系列推荐,尽量按国家标准。工作中我们经常采用线性校准,因为线性方程最为简洁。 标准曲线需要几个数据点,是由所检测组分的浓度范围、分析仪响应特性、干扰因素、浓度与检测信号响应类型有关的。 3个点:对于一些低浓度,特别是微量分析,并且浓度范围不是很大的,检测器响应可靠,背景干扰非常小的,则可以选用较少的工作点就行,一般有3个浓度点就足够了,有些甚至可以只用一个浓度点就行,另一个点直接用坐标原点。 4~6 个点:对于平时测量的样品浓度范围较宽,并且检测器响应不完全是一次曲线(直线)的,可以采用二次曲线或分段校正方式,以减少数据偏差,这种情况就需要多用几个数据点,如果不分段,数据点有4~6 个就够用。但如果是分段校正,则每段需要至少两个数据点。而对于一些样品无浓度范围规律的,特别是一些检测机构,如果分析仪的检测响应可靠,环境因素影响少的,可以做校正曲线。若是环境因素影响大的,则不必做校正曲线,而采用标准加入法或标准加入-一次稀释法反而简便一些。 实际上,一般是做五个点(不包括零浓度);一般以检出限的5~10倍为第一个点,以后根据1倍(或接近一倍)递增,最高浓度是最低浓度的10~20倍为宜。当然,要根据仪器的灵敏度来调整。 标曲R值是几个9才合格? 实验室应按检测标准(方法)的要求使用标准溶液或标准物质建立标准曲线。所用标准溶液或标准物质应覆盖被测样品的浓度范围。检测标准(方法)如无具体的要求,至少使用5个标样(除空白外)建立线性标准曲线,每个点重复测定1-3次,对于筛选方法,线性回归方程的相关系数不应低于0.98,对于确证方法,相关系数不应低于0.99。其实〉0.99是判断是否为线性相关的一个标准,实际应用中线性 〉0.999才是比较理想的。线性在0.99到0.999之间的监测结果只用接近最高浓度一半(中间浓度)的位置才比较准确,如果线性大于0.999的话,在整个线性范围内都会有一个比较满意的结果。如果检测的线性不好,可以减少标准的覆盖范围,将标准的浓度调整到待测样品浓度附近,这样结果也是非常准确的。例如,样品的浓度约20ppb,但在0~50ppb范围建立标准曲线,但线性非常不理想,这时可以将标准范围调整到15~25ppb之间,作五个标准。标准曲线建立后应在样品的检测中消除空白造成的影响。高于接受限的试剂空白表示与空白同时分析的这批样品可能受到污染,检测结果不能被接受。标准曲线建立后,必要时应在分析样品前加标,添加物浓度水平应接近分析物浓度或在校准曲线中间范围浓度内,加入的添加物总量不应显著改变样品基体。标准曲线有效期到底规定:(1)标准曲线属于实验室质量控制的范围,按照《实验室资质认定评审准则》中结果控制的要求:定期使用有证标准物质(参考物质)进行监控和/或使用次级标准物质(参考物质)开展内部质量控制。(2)准则中并未对“定期”进行规定,所以如果“定期”,就根据实验室的实际情况来定了。(3)既然准则上没说明,那根据一般的分析教材,当实验条件(包括药剂、人员、仪器等)发生变化时,最好重新制作标准曲线。一般来说仪器如果长期使用,并经过检定,是处于稳定状态,而人员药剂的变化往往会较大。如果说每次换个人操作都要换曲线,那工作量就太大了。每次开机都必须做标准曲线?从药品检测的要求来说,因为:1、每天所配的流动相都会有所不同,导致出峰的时间都会有一定的差异,峰面积相应都有所差异。2、检测器光能也在不断的衰弱,因此其每天的相应值也有所不同,其峰面积也有差异。基于以上原因,原则上应该是每天都要进行标准曲线校正的。标准曲线不要每次都做,但是每次必须进标准品样品;因为每次你的流动相和原来的不可能完全一样,同时仪器的状态也在变化,所以不同批次间的保留时间是不一致的,所以你必须用随行标准品来定位与定量;标准曲线不一定非要过原点,只要线性好就可以了。因为做空白的话基本上不会过原点。过原点是强制过的,实际曲线可能不过,就会造成误差。不过原点是因为有系统误差。不过系统误差的话,大家都误差也就抵消了,没有太大影响,针对是否过原点(0,0)问题,可以从原点(0,0)代表的意义来考虑:即所测组分浓度为零的时候,信号响应值(液相色谱也就是峰高或者峰面积)是否也为零。通常来说色谱分析是属于这种情况的,因而可以把(0,0)点作为一个浓度水平计入标准曲线(甚至不需要真的去配一个浓度为零的标准溶液来进样),这也是单点法定量的一个依据;标准曲线的线性范围线性范围,主要从相关系数r看,一般要求r大于等于三个九。之前做实验有时候浓度高时,线性不好,高浓度点不在标准曲线上,而是在标准曲线的下面,而且离拟合的标准曲线比较远。遇到这种情况,标准曲线的线性相关系数就很差,有时候才一个九,,如果自己用手动拟合的话,用平滑的曲线去连接所有点的话,你就会发现,如果在线性范围内,连接起来就是直线,如果超出了线性范围,连接起来就是一条弯曲的曲线。标准曲线的相关系数的有效数字该如何保留呢?GB5750.3-2006 8.2.7项有如下规定:校准曲线相关系数只舍不入,保留到小数点后出现非9的一位,如0.99989→0.9998。如果小数点后都是9时,最多保留小数点后4位。小结标准曲线法有一定的优势,也有一定的缺陷,它特别适合于大量样品的分析。但由于每次样品分析的色谱条件(检测器的响应性能,柱温,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。此外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),而实际样品的组成却千差万别,因此必将给测量带来一定的误差。
我是学有机化学的。目前目标分子的单晶结构对我非常重要。请大家推荐单晶结构分析的软件!当然,免费的最好![em51]
在分析化学实验中,常用标准曲线法进行定量分析。现在做标准曲线的方法是五花八门,有坐标纸绘制法,计算器算法,电脑软件绘制法和分析仪器自动计算的。你在分析中一般都使用的是什么方法,在绘制曲线时有什么好的经验,诀窍和感受,在这里和大家分享下吧!
写在前面的废话: 去年编写的一个分析化学学习软件,因为要参加省和国家的多媒体大赛,所以在论坛中只是作了演示的介绍。软件介绍见(含动画演示): http://bbs.instrument.com.cn/shtml/20100627/2634043/ 参赛结果:评了个省级二等奖,国家级落选了。http://simg.instrument.com.cn/bbs/images/brow/em09508.gifhttp://simg.instrument.com.cn/bbs/images/brow/em09508.gifhttp://simg.instrument.com.cn/bbs/images/brow/em09508.gif 评个什么奖倒也无关紧要,反正也不需要评职称了。一个花了很长时间心血的作品,囚在自己的电脑中“孤芳自赏”,也觉得无趣,就共享给大家吧。不求你评价,只图你有用,就当是在论坛中摆个“地摊”,搞了个“个展”,让那些支离破碎的分析化学基础知识能唤起大家渐已淡忘的大学回忆…… 下面是《软件使用手册》:http://ng1.17img.cn/bbsfiles/images/2017/10/20110607073547712_01_0_3.swf 如果要下载《软件使用手册》,请下载附件。《软件设计手册》就不介绍也不上传了。(软件看下帖)

毛巾浴衣标准中吸水性标准中有两个检测方法,标准中没有要求用哪个方法检测,这个大家怎么操作?[img=,309,33]http://ng1.17img.cn/bbsfiles/images/2017/10/201710051626_01_2154459_3.png[/img][img=,690,134]http://ng1.17img.cn/bbsfiles/images/2017/10/201710051626_02_2154459_3.png[/img]
热分析ASTM标准找齐了,作为圣诞礼物送给大家!!分两块,大家分别去下面的两个链接(两个关联的帖子)去下载吧,不要积分的。http://www.instrument.com.cn/bbs/shtml/20051128/286137/http://www.instrument.com.cn/bbs/shtml/20071221/1102830/
请问专家国家标准中有没有分析聚合级丙稀中微量的一氧化碳、二氧化碳的方法
各位兄弟姐妹,大家好。实验中遇到不顺,真诚的请教大家。我做的是苯酚羟基化反应,产物中有苯酚,苯二酚(邻,对),苯醌,焦油,用安捷伦1100液相色谱分析,外标法计算选择性和转化率。但一直以来,计算出的结果老是差强人意。偶尔算出一个好的结果,但是一重复又不行了,有时候重复做几次都不一样,很是郁闷。最近又老算出来选择性超过100%。开始怀疑稀释误差的问题(样品是稀释以后去测的),但现在操作的时候非常小心了,怀疑标准曲线的问题,但是重新做了好几条了。但结果就是很奇怪,自己想了想:1.因为液相色谱仪是公用的,所以特意买了专用的色谱柱。但经常测的时候发现柱压不一样,是不是因为这个影响了峰面积,进而影响了结果?柱压的影响这么大么?2.是不是外标法就不准确?该用单点校正法?之前觉得每次测样前都配一个标样有点麻烦,所以没用。3.还是别的什么原因?我用的柱子是ZORBAX SB-C18, 用的甲醇:水=3:7,每个峰都分的很开。取样后离心去掉催化剂,再定量稀释然后去测的。因为分析的问题有时候做实验都怕是白做,很着急,希望好心人和懂的高手给予指点,不胜感激,谢谢了,祝你们学业事业有成。
最近做实验用分光光度法分析 烦人呀 每分析一批样都要做标准曲线(书上这样说的) 曲线做出来了 时间也所剩不多 我想请问大家:可以不每次都做标准曲线吗?我还有一个问题感到很郁闷的是 我做萃取实验 每一次分析的原始溶液的浓度都不同 计算的萃取率也乱七八糟的 因为初始的浓度不一样 我想都校正到一个值上 但是找不到依据呢。请大家帮帮我呀 我都急死可 实验做不走呢
问下大家Gb/T223 中化学分析用的标准液体可以买吗? 比如我用的标准GB/T223.5 GB/T223.59 GB/T223.23 GB/T223.26 GB/T223.11 这几个标准中配置的 标准溶液 可以买到吗 ?买GBW(E)可以吗 CMA计量认证
大家都来说说用什么软件搞元素分析后的成图我用coredraw和graph
样品中的铬含量大概3%.我配制两个标准系列.低浓度:0,2,4,6,8,10mg/l(将1000 mg/l标准储备液稀释10倍,因要测别的样,稀释的100mg/l的标准溶液放置三天才配标准系列)高浓度:0,10,20,30,40,50mg/l(直接用1000 mg/l标准储备液配制)两个系列测的结果由低浓度测出3.6%,高浓度测出3.0%,我分析的原因是低浓度系列所用工作液放置了三天,请大家帮我分析下原因.谢谢了
问下大家Gb/T223 中化学分析用的标准液体可以买吗? 比如我用的标准GB/T223.5 GB/T223.59 GB/T223.23 GB/T223.26 GB/T223.11 这几个标准中配置的 标准溶液 可以买到吗 ?买GBW(E)可以吗 CMA计量认证
您好!我想向专家咨询硫醇甲基锡热稳定剂中有机锡和硫含量的标准分析方法?我查了很多资料都无结果!谢谢!
各位同行: 在农药残留分析中,一个讨论较多的话题就是关于基质效应的问题,一般采用添加基质的标准品来补偿基质效应,但是在具体的配制过程中,现在尚为有一个统一规范的标准,请大家都来讨论一下,各自在平时的实验中,是怎么样来操作的! 此贴只为抛砖引玉,希望大家踊跃讨论,有好的操作,我们一起学习!
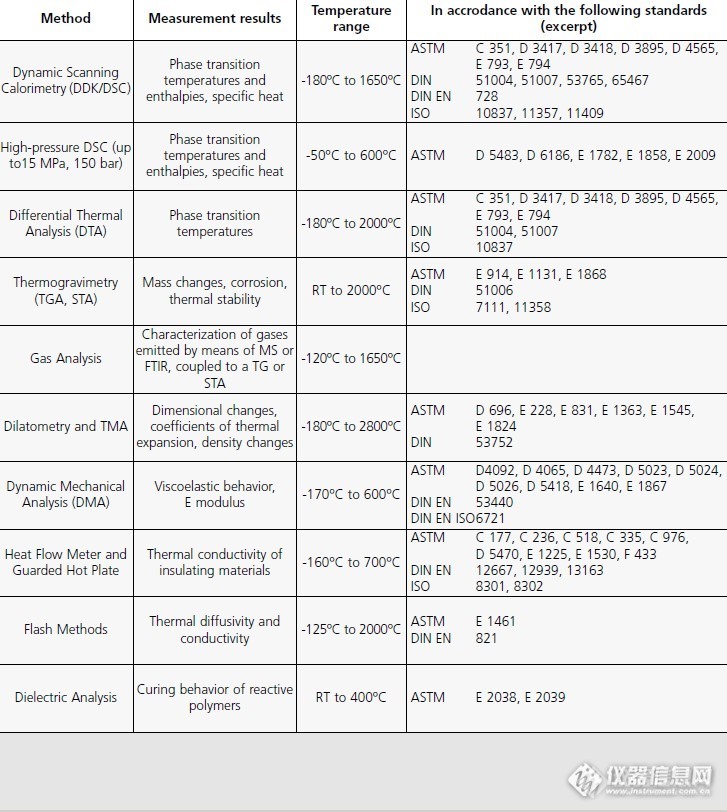
今天整理资料,看到一份耐驰(NETZSCH)公司的技术宣传文件,其中有一张表,内容是他们仪器可适用的国际上一些标准测试方法。我觉得这对各位使用热分析仪应该很有参考意义,贴上来供大家分享。http://ng1.17img.cn/bbsfiles/images/2013/07/201307262208_453972_1633752_3.jpg
木棉是标准的纤维名称吗?大家有检测遇到过木棉的成分分析吗?
在定量分析中, 大家都用什么溶剂来配标准溶液. 有人用MeOH, 有人用DMSO, 有人用DMF.每个人都有一套说法. 说一说你都用什么溶剂配标准溶液?
现在急需一份固体甲醇钠的分析方法,企业标准或行业标准都行。希望大家帮帮忙







 我要推广仪器
我要推广仪器
 下载APP
下载APP