SP-3520AA[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计为什么没有扣除标准空白的功能,曲线不扣除标准空白的吸光,对吗?还有这个型号的仪器做样品时,减的是样品空白的浓度,为什么这台仪器和别家的不一样?这样减到底对不对?
火焰法测量铅时,用标准空白溶液校零,但是测量的时候,吸光度还是不会为零(比如0.001)。作图或者拟合曲线的时候是用标准溶液的吸光度减掉标准空白的吸光度吗?还有,测量未知样品的时候也会有样品空白,样品空白的吸光度如何处理,是要扣除,还是按照零计算。软件上直接告诉浓度,但是理论计算到底按照哪种方式计算是正确的。
最近在做Fe 元素,用的火焰法,仪器是pe900t ,波长248.3nm, 仪器开机预热够了,灯也预热了,设置好参数后,打开连续图谱,发现上边吸光度值为-0.0100,点过后,用超纯水清洗系统后,看吸光度值依然为负值,让后检零后,测试标准空白溶液(0.1mol /L 硝酸),发现吸光度为-0.0089,很好奇,为什么空白吸光度会为负值?然后看了灯能量为33,会不会是我的灯能量不够了?应该换灯了,还是空白低于了检测限,或者还有其它原因造成的?
做标准曲线,标准空白的吸光度有时候是负值,怎么回事呀?[em0804]
AHMT法测甲醛含量中,标准曲线拟合时的纵坐标吸光度需要减掉空白吸光度吗
最近用火焰AAS测Cr时,配了0,0.5,1ppm三个点做标准曲线,线性还行(0.998左右),但标准空白的吸光度为-0.005左右,为何会这样?虽然测定结果落在线性范围内,但不知道测定的准确性如何?请各位高手指教一二.

请问大家,水样颜色较深,做了色度补偿,减色度补偿的吸光度后,还需要减空白吸光度嘛? 我认为是不用再扣除空白的,因为色度补偿的样品只是没有加显色剂,其他试剂的影响都已经扣除了,再减空白的话就重复扣除了。 但是我们实验室内部无法统一观点,请教一下大家是怎么操作的? 苯胺类标准 GB 11889-1989[img=,690,161]https://ng1.17img.cn/bbsfiles/images/2020/08/202008121234425275_5197_3079646_3.png!w690x161.jpg[/img] 六价铬标准GB 7467-1987[img=,690,233]https://ng1.17img.cn/bbsfiles/images/2020/08/202008121237081623_6224_3079646_3.png!w690x233.jpg[/img]
测环境空气臭氧时,标准曲线第六个点吸光度值最高,是0.903,可是空白样品的吸光度值应该不高于0.903,可是我能测空白样吸光度是0.977,这个是怎么回事?
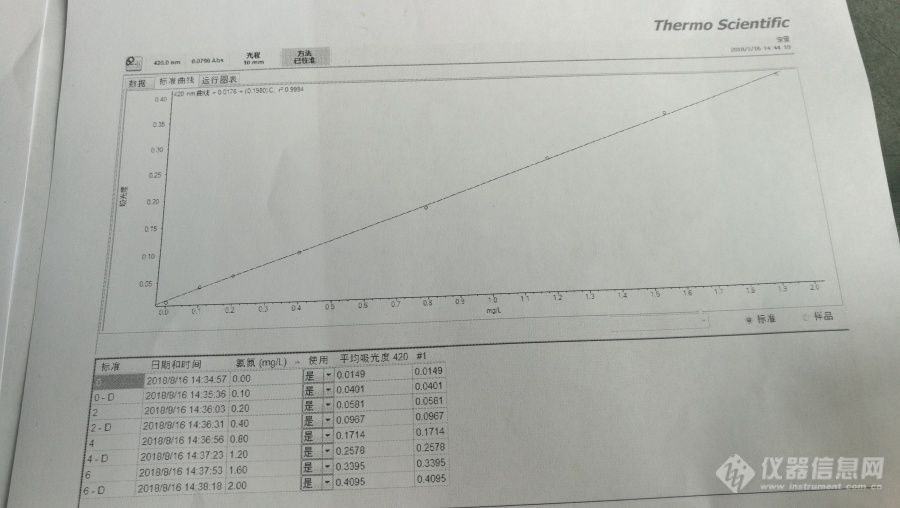
[img=,690,389]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161633404032_1450_3294036_3.jpg!w690x389.jpg[/img]如题,我们用的机器是赛默飞 Evolution 201 ,10cm比色皿,测完直接就出来这样的结果。曲线没有扣除空白,并且吸光度也是根据浓度回归,不知道直接这样可以不,另外,第一次做氨氮,这个曲线还行吧??传上去好像看的不是很清楚,曲线是y=0.198x+0.0176 r2=0.9994
我是做生活饮用水的检测,参考国标5750-2006,现在很多分光光度计的软件可以自动扣除空白,即用空白调零,看到有人用分光光度计做实验时,不管国标中以什么为参比,都用空白调零,不知道这对结果影响大不?当然她有做空白平行。我的理解是按国标来讲,以什么为参比就以什么为调零,结果按步骤来,数据处理也不扣除空白。如果说空白对结果影响不大,最终结果都要扣除空白,那国标为什么要说以什么为参比,直接都用空白调零就好了,难道仅仅是为了测试看空白是否符合要求,然后数据处理都要扣除空白?如果我测的空白符合要求,我数据结果也不扣除空白,对数据结果有影响不?望前辈们指教下
我想问下紫外分光光度计法单光束,测定吸光度,扣除空白溶剂(0浓度)点校准吸光度,在用校准吸光度绘制标准曲线时,是否再次带入(0,0)点去计算标准曲线的斜率和截距呢?就是扣完空白后,直接用1,3,5,7,9点位的吸光度去计算校准曲线,或还是得把0,1,3,5,7,9校准后的吸光度代进去计算曲线
采用标准加入法测定基质复杂的样品与采用标准曲线法测定一样,都是应该扣除试剂空白的,尤其是使用的硝酸含有较高待测本底的情况下,这一点应该是毋庸置疑的。总不能把试剂本底也算到样品中去吧?问题是该如何扣除掉试剂空白值?迄今为止,尽管经过多方查询,我始终未能得到较为权威的计算方法。把道听途说的方法粗略合计一下,再结合自己的一些想法,我觉得可以把归这些归纳为三种方法:1.把试剂空白的吸光值从添加物系列以及样品的吸光值中扣去,然后代入校准方程计算出样品的浓度值;2.把试剂空白的吸光值代入校准方程计算出浓度值,然后将该值从样品的测定值中减去;3.采用标准曲线法测出试剂空白的浓度值,然后将该值从样品的测定值中减去。下面试分析这三种方法之优劣:第1种:这种扣除试剂空白的方法跟火焰标加法相类似,但我认为这样做不妥。因为之所以采用标加法,往往是因为样品的基质干扰待测元素的测定,而试剂空白中没有样品,自然就不受基质的干扰,因此试剂空白中试剂的吸光值与其在样品中时是不同的,故不应该将在外测得的吸光值从样品内扣去。第2种:虽与第1种看上去不同,但本质是一样的,它也不能回避试剂空白没有受到基质干扰的影响。第3种:正是由于试剂空白不受基质干扰,所以可以直接采用标准曲线法测出,这个值是可信的,而采用标准加入法测出样品的含量也是可信的。将后者减去前者,从而得到样品待测元素的结果是可信的。我认为该法值得推荐。曾参看某贴,认为“ 如果需要进行“空白”校正,就需要另外制备一组样品:他们的组成,除不含待测物外,其余都应与被分析的样品相似。同样应用标准加入法,从吸光度-浓度的关系,求出浓度坐标上的截距,作为“空白”校正浓度。”“其方法是:用于校正的“空白”浓度等于空白标准溶液的计算浓度乘以上述两个校正曲线的斜率比(标准加入法曲线斜率/标准曲线发斜率)。”沉思良久,不明其意,故不敢苟同。我以为扣除标准加入法测定结果的试剂空白值不是一个可以忽略的问题,它与我们采用标准曲线法测定需要扣除试剂空白一样的意义。否则,我们采用后者测定也就不要扣试剂空白了。不,干脆连做试剂空白都不要做算了!这样可以吗?我看不行。以上讨论仅限于适合采用标准加入法的样品测定过程,那些电离干扰、光散射和分子吸收产生的衰减干扰以及光谱干扰造成标准加入法不能克服的测定过程不在本讨论之列。
做锌的标准曲线 ,0 0.1 0.2 0.4 ,0的吸光度是负的0.0034,这个是什么原因呢 还有样品空白也出现了负值,负的0.0153,这种情况正常吗
PE AA700测食品中的铅,做标准曲线的时候吸光度特别高,标准曲线线性还是很不错的能达到3个9,从校准空白开始吸光度就特别高达到0.3甚至更高,于是整个系列都特别高,最后一个点都1点多了,之前做都没出现过这种情况,就是最近的现象,大家说这是怎么回事呢,郁闷呐,会是针没调好的问题吗,十分着急,渴望大家的回复!!!谢谢
1、这两天测监督性废水,用高锰酸钾氧化-分光光度法测总铬,发现消解空白达40-50 ppb,2次平行挺好2、以前用过硫酸铵作氧化剂时,有时消解空白有检出,有时没有,同批次样品的平行不好(包括消解空白)、不同批次的标准点样品平行不好我想问的是,如果是试剂或水的问题,应该不至于消解空白的平行不好,或者不至于测出消解空白那么高,会是什么原因呢?疑问:纯水机制的纯水,三价铬的浓度大概多少?有人测过吗?谢谢!注:消解空白,加尿素,再滴加亚硝酸钠后,是无色、澄清的样品,按理说不存在色度、浊度干扰,显色后有明显的紫红色物质产生,颜色深浅与相近标准点溶液的相当初步排查表明:磷酸带来了三价铬污染,但空白的吸光度重复性很好,可以通过扣除试剂空白的方式降低干扰带来的影响;所用硫酸无三价铬本底
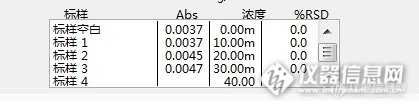
比如,第一次标准空白三次吸光度分别为 0.0036 0.0024 0.0027;做完样品后第二次分析分别为0.0038 0.0022 0.0015。
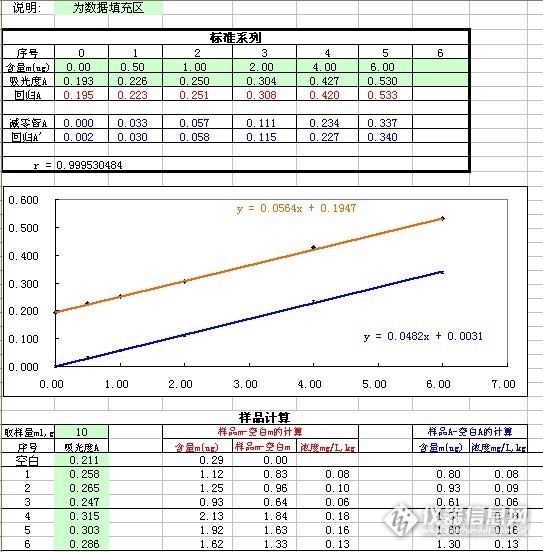
GBT/T 160.10 双硫腙分光光度法测工作场所中铅含量,红色的是样品铅含量-空白铅含量,蓝色是样品吸光度值-空白吸光度值,最后的计算结果不一致,应该选哪种扣空白方式?http://ng1.17img.cn/bbsfiles/images/2013/05/201305171012_440488_2694188_3.jpg

(1)仪器型号:AA-7003A(2)背景扣除方式:氘灯(90)(3)分析模式:石墨炉(4)待测元素名称:Sn(5)标准样品浓度:0ug/L(2%硝酸)(6)检查过程:打入20uL的硝酸进入石墨炉测量,吸光度很高(7)测试结果和图谱:测量过几次有0.0456、0.1256、0.0756、0.1554等等,所用的石墨管是新的,测定Cd,打入同样的量跟试剂,所测吸光度是0.0120、0.0101。如图是测量的条件,还有吸光度的走向图(8)疑问和求助内容:换了一次仪器内部的通水管路之后,测量Sn这个元素的空白吸光度就变得很高,测量Cd、Pb这两个元素倒没有很高。在换之前测所有的元素空白值都挺正常的,换了之后就唯独Sn这个元素的空白吸光度变得很高(前后测Sn所用参数一致)或许是因为灯的原因?Sn元素灯坏了?[img=,690,264]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021050584855_1011_3983958_3.png!w690x264.jpg[/img][img=,690,592]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021051087790_4193_3983958_3.png!w690x592.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021051214750_2464_3983958_3.jpg!w690x517.jpg[/img]
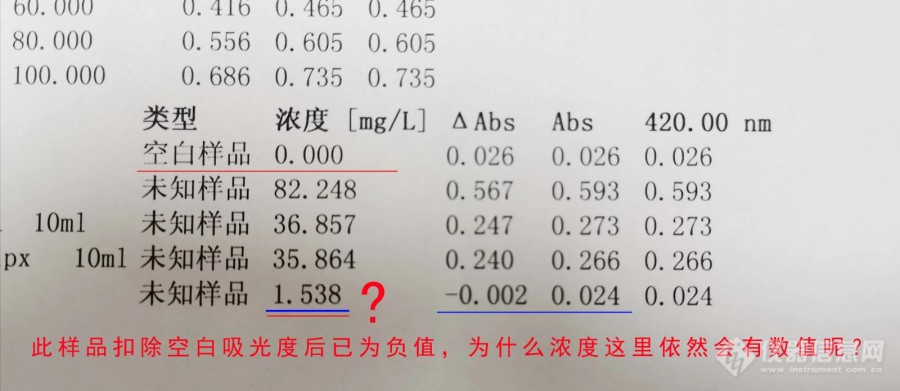
[img=,690,299]https://ng1.17img.cn/bbsfiles/images/2023/07/202307191011421682_4455_5273478_3.jpg!w690x299.jpg[/img][size=18px]如图,使用的普析TU1900分光光度计,设置空白样品后做样,为什么样品扣除空白的吸光度后,明明已经是负值了(0.024-0.026=-0.002),但浓度那里还有数值??空白(0.026)浓度为0,样品(-0.002)浓度却为1.538??是仪器设置问题吗?[/size]
关于扣除方法空白的疑惑,我做的半导体的痕量分析。1、如果以超纯水加标做标准曲线用标加法做的,将方法空白作为未知样品来做样,那么我待测样品如何去分析?在标准曲线的界面上选择减去方法空白吗?工程师说有些样品脏就减去,但是有些样品基体较干净就不需要减,减去后会出现小于零。因为怕空白被污染,但是减与不减的结果有差异的,我们做检测的不是要求数据要准确吗?2、如果以方法空白加标做标准曲线也用标准加入法做,将方法空白作为曲线零点来做,那么我待测样品如何去分析?在标准曲线的界面上还是选择减去方法空白吗?工程师依然说有些样品脏就减去,但是有些样品基体较干净就不需要减,理由还是如上。这样的两种说法,第一个的前半部分可以接受,可减去方法空白,但是后面的说法就不明白了。也有朋友说标准加入法不需要减去空白。哪位高手可以解说下吗?谢谢
耶拿650P 为啥空白吸光度和标曲吸光度一样啊?昨天都好好的,今天就不给力了,刚开始玩这个玩意儿!我测定是铜和铅,标准使用液浓度是20ppb空白吸光度为0.01037 然后曲线的分别是0.01041 0.01037等等,反正差别不大,浓度分别是 0 4 8 12 16 20ppb
我才开始学测食品中的二氧化硫,最近几次都不成功,做标准曲线做不成,空白的吸光度在0.3左右,以空白校零后,测得的标准溶液的吸光度都在0.0几左右,这样的结果对吗?我测得盐渍姜的二氧化硫含量在50到80mg/kg之间。这样的结果合理吗?有没有大侠们也做这个的。给点数据参考参考呗!先谢谢各位!
耶拿的原子吸收,测锌时用超纯水调零,标准空白的吸光度-0.107,为什么? 用其它正常的铜标准溶液的标准空白,也是同样的结果(测铜时,一切正常)
在测定蔬菜中Cr,做标准曲线时,测得的标样空白吸光度为0.31(空白标样只有水和基改剂),而标样系列中的4ppb浓度的吸光度只有0.17,8ppb的吸光度为0.36,难道空白标样中不能加入基改剂吗?空白标样中不加基改剂,只进水的时候,吸光度为0.02,因此我想引起吸光度增大的只有是基改剂了。请帮帮忙!
原吸测铅镉问题 求教:原吸测Cd 和 Pb 的问题 我们单位去年买的原吸 日立-2700 石墨炉法,我们主要做的是原粮类。刚买回来的时候做铅和镉曲线很容易就能达到0.995以上,但是今年做的时候曲线的线性关系就不咋好了, 比如做镉 :0ng/ml 的吸光度为 0.0006 1.00ng/ml 的吸光度为 0.0497 2.00ng/ml 的吸光度为 0.1013 4.00ng/ml 的吸光度为 0.1888 6.00ng/ml 的吸光度为 0.2577 8.00ng/ml 的吸光度为 0.3173 最后两个点跟前面就不成比例了,我开始以为是最后两个点的标液被污染了,但是我重新用1+1硝酸泡过后还是出现同样的问题,包括铅的曲线也是一样,最后两个点和前面的始终都不成比例关系。 还有就是我们做样品的时候,比如同一根比色管倒出的未知样品上机测定,铅的空白很高,基本上都比样品的空白要高,所以把样品空白扣除了以后样品的浓度都成负值了,而且带的标准物质测得值也是负的;但是测镉却是正常的,而且测得标准物质的浓度也非常接近。因为这个问题,做了很多次都是这样,包括重新配置硝酸泡瓶子,重新稀释标液也做过。 我们的样品是采用微波消解,CEM公司的微波消解仪。消解加的是1+1硝酸8ml,称量0.49g左右,消解完后没有进行赶酸, 求各位前辈多多指教。谢谢了哟
邻菲啰啉分光光度法测溶液的铁含量,溶液本身是黄色的话,空白怎么做?1.我如果用蒸馏水做空白,空白的吸光度是0.0502.我如果用待测溶液做空白,除了菲啰啉不加外,别的都加了,显示的吸光度是0.133那我到底该怎么做空白呢,谢谢指教~
http://ng1.17img.cn/bbsfiles/images/2017/01/201701121449_01_3157743_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/01/201701121454_01_3157743_3.png仪器型号:AAS-240Z前几天测铅都挺正常的,昨天开始测完标准曲线之后测样品结果都是负值且不同样品结果差不多,今天重新再测,标准曲线吸光度和标准空白吸光度差不多,不成线性关系。重新换了稀释液结果一样。突然之间变成这样,我有点措手不及,不知道是不是灯的原因,优化了灯,调换灯的位置以及调整灯的位置都没有用。http://ng1.17img.cn/bbsfiles/images/2017/01/201701121456_01_3157743_3.png这是进样视频http://ng1.17img.cn/bbsfiles/images/2017/01/201701121458_01_3157743_3.png之前都正常的,没有调动过哪儿,不知道是不是灯的原因!请大家指点迷津啊!
一级 二级 吸光度(254nm,1cm 光程)≤ 0.001 0.01 这是试验用水有关吸光度值的标准.但我不知道它是什么为空白的(用什么来调零)?是以空气吗?[em09509]不好意思!没说清楚,我用的是流动比色皿!这可该怎么办呢?
制作龙葵素标准曲线 没有用空白调零 试验组中龙葵素吸光度可以用试验组减去空白组的吸光度算么
我这次做氨的标准曲线时,用的是靛酚蓝分光光度法,测的吸光度普遍偏高,空白氨的吸光度为0.199,当氨含量为0.1微克时,吸光度就达到了0.65了,而且在氨含量从0.1-5微克之间时,随着氨含量的增大,吸光度随着增大,但是当氨含量大于5微克以后,吸光度反而在开始减小了,这种现象很奇怪,我都是按照国标方法来配的溶液,操作方法来是按照标准来的,不知道为什么吸光度会这么大,而且会出现先增大后变小的现象,希望大家能帮我解释下。还有就是不知道是不是跟我先加的标液,后加的吸收液这一顺序有关系。急切想找到答案,希望大家帮帮我这个菜鸟。非常感谢。







 我要推广仪器
我要推广仪器
 下载APP
下载APP