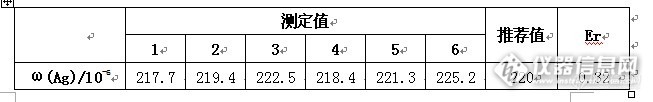
火焰原子吸收光谱法测定矿石中银含量【摘要】:本方法用盐酸和硝酸溶解样品,硫脲络合银,测定矿石中银含量,检出限低(0.006ug/ml)、精密度好(RSD为1.3%)、准确度高(Er为0.32)。【关键词】:火焰原子吸收分光光度计矿石银盐酸硝酸银在自然界中主要以硫化物的形式存在,大部分是伴生在铜矿、铜铅锌多金属矿、铜镍矿和金矿床中,单独存在的银矿物如辉银矿(Ag2S)少见。在开采和提炼铜、铅、锌、镍、金时,含银达5g/t即可综合利用。银的边界品位为40g/t。银含量的测定是评价银矿石和含银副产矿的首要工作。目前,矿石中银含量的测定方法有:分光光度法、发射光谱法、火焰原子吸收光谱法、电感耦合等离子体质谱法等。火焰原子吸收光谱法,因方法简便和适合测定微克级含量银,而常被用于矿石中低含量银的测定,此法一般将样品预处理为强酸或氨水等介质,其不足之处是溶液中强酸或氨水的浓度较高,易对原子化器产生较大的腐蚀作用。本文根据硫脲能与银形成可溶性稳定络合物的特点,尝试了样品经王水分解,硫脲提取,空气-乙炔火焰原子吸收光谱法测定矿石样品中的银,结果与推荐值相符。1、实验部分1.1、主要仪器和试剂仪器:TAS-990原子吸收光谱仪(北京普析通用仪器有限公司)、分析天平、锥形瓶、容量瓶、电热板试剂:盐酸、硝酸、硫脲、纯净水标准物质:多金属矿石成分分析国家标准物质GBW071631.2、TAS-990原子吸收光谱仪工作条件http://ng1.17img.cn/bbsfiles/images/2013/09/201309280943_467978_2352694_3.jpg1.3、标准曲线绘制用1.0mg/ml银标准溶液逐级稀释,配置系列标准溶液。http://ng1.17img.cn/bbsfiles/images/2013/09/201309280944_467979_2352694_3.jpg1.4、样品预处理1.4.1、含量与取样量关系表http://ng1.17img.cn/bbsfiles/images/2013/09/201309280944_467980_2352694_3.jpg1.4.2、样品预处理按照上述表格中大概含量取样品GBW07163于150ml烧杯中,用少量水润湿。加入10ml盐酸,在低温电热板上煮沸20min,加入5ml硝酸,继续煮沸至黄烟消失。将溶液蒸至近干,取下冷却至室温 ,用少量水冲洗杯壁,加热溶解盐类。取下冷却,[/s
g/b 3884.2-2000 中使用原子吸收测定铜精矿中银,银标准溶液使用的是20%的盐酸介质,盐酸介质达到20%以上才不会有氯化银沉淀,为什么在样品处理中,最后定溶的盐酸介质都是10%,难道不会有氯化银沉淀吗?对结果不影响吗
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=105188]火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定纯铜中银[/url]
各位大侠帮帮忙!我们是矿物分析,以前是[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定银后,再测定铅。现在求一连续测定方法,在测定银的同时,可测定出铅或铜.
各位,我用的是火焰原子吸收分光光度计,用来测纳米银中银含量,用电导仪测大概在150ppm左右,我想问的是具体怎么操作,还有标准曲线配置有什么要求,多久就要重新做标准曲线?谢谢啦

火焰原子吸收法测定铜精矿中的锌的含量1. 前言论坛上看到有坛友为用原子吸收测定锌含量抓狂,正好前段时间做了方法验证,现在分享一下我们的实验过程,个人浅见,不足之处,还请各位专家老师指正。样品经四酸消解,在稀盐酸介质中,以空气-乙炔火焰,用原子吸收光谱仪测定元素锌的吸光度,扣除背景吸收,按标准曲线法计算锌元素的含量。经标准物质实验验证,方法的回收率在95%~105%;精密度RSD在3%以内,完全可以满足质量控制要求,现已用于实验室矿物中锌含量的测定。2. 试剂2.1 盐酸(分析纯,广州试剂厂)2.2 硝酸(分析纯,广州试剂厂)2.3 高氯酸(分析纯,广州试剂厂)2.4 氢氟酸(分析纯,广州试剂厂)2.5 1000µg/ml锌标准储备溶液(国家有色金属及电子材料分析测试中心生产)3.仪器设备3.1 电子天平http://ng1.17img.cn/bbsfiles/images/2014/11/201411252131_524609_1657564_3.jpg3.2 特氟龙试管http://ng1.17img.cn/bbsfiles/images/2014/11/201411252133_524610_1657564_3.png3.3石墨消解炉http://ng1.17img.cn/bbsfiles/images/2014/11/201411252135_524611_1657564_3.jpg3.4 瓶顶移液器http://ng1.17img.cn/bbsfiles/images/2014/11/201411252136_524612_1657564_3.jpg3.5 计时器3.6 容量瓶3.7 原子吸收分光光度计http://ng1.17img.cn/bbsfiles/images/2014/11/201411252153_524613_1657564_3.jpg4. 样品消解4.1 准确称取0.2~0.25g(精确至0.001g)样品于Teflon试管中,并将试管置于试管架上;4.2 用瓶顶移液器准确向每根试管中加入1.5mL的浓硝酸和1.0mL高氯酸,轻摇试管架使样品充分润湿,待其稳定约5分钟;4.3 将试管放入已恒温至115±5℃的石墨消解炉上,加热消解样品5分钟;4.4 将试管取下并放在试管架上,冷却约5分钟;4.5 用瓶顶移液器向每根试管中准确加入2mL的氢氟酸,再返回至115±5℃的石墨消解炉上加热消解30分钟;4.6 将试管转移至185±10℃的石墨消解炉上继续消解2.5小时,此时溶液应蒸发至近干;(注:如果溶液被完全蒸干,则此样品应重新称量并消解)4.7 取下试管,待其冷却5分钟;4.8 向每根试管中加入5mL50%盐酸,再将试管放回至185±5℃的石墨消解炉上,加热15分钟;4.9 取下试管冷却至室温,用5%盐酸,定容至100ml反复摇晃3~5 分钟,使溶液充分混匀;静置并作记录,待仪器分析。5. 测定5.1 仪器参数http://ng1.17img.cn/bbsfiles/images/2014/11/201411262150_524756_165
[size=32px][b]关于发布《水质 锑的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法》等五项国家环境保护标准的公告[/b][/size] 为贯彻《中华人民共和国环境保护法》和《中华人民共和国水污染防治法》,保护生态环境,保障人体健康,规范生态环境监测工作,现批准《水质 锑的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法》等五项标准为国家环境保护标准,并予发布。 标准名称、编号如下。 一、[url=http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/201911/t20191101_740413.shtml]《水质 锑的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法》(HJ 1046-2019)[/url]; 二、[url=http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/201911/t20191101_740415.shtml]《水质 锑的测定 石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法》(HJ 1047-2019)[/url]; 三、[url=http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/201911/t20191101_740416.shtml]《水质 17种苯胺类化合物的测定 液相色谱-三重四极杆质谱法》(HJ 1048-2019)[/url]; 四、[url=http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/201911/t20191101_740417.shtml]《水质 4种硝基酚类化合物的测定 液相色谱-三重四极杆质谱法》(HJ 1049-2019)[/url]; 五、[url=http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/201911/t20191101_740419.shtml]《水质 氯酸盐、亚氯酸盐、溴酸盐、二氯乙酸和三氯乙酸的测定 [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法》(HJ 1050-2019)[/url]。 以上标准自2020年4月24日起实施,由中国环境出版集团有限公司出版,标准内容可在生态环境部网站(http://www.mee.gov.cn)查询。 特此公告。[align=right] 生态环境部[/align][align=right] 2019年10月24日[/align] (此件社会公开) 抄送: 各省、自治区、直辖市生态环境厅(局),新疆生产建设兵团生态环境局,各流域海域生态环境监督管理局,环境标准研究所,各标准承担单位。 生态环境部办公厅2019年10月29日印发
铅的EDTA滴淀法大家都是知道的。大家有没有铅银矿中铅的原子吸收火焰分析方法。在这里先谢谢大家啦!!如果有锌的一起发来,再次谢谢啦。。
我是个原子吸收菜鸟级人物,却碰到个最复杂的事情,纵观版面,很少有提到在矿物质原料中铅、铬、镉的测定,希望专家们来讨论下我们是饲料企业,需要用到硫酸铜、硫酸锌、石粉(90%以上碳酸钙成分)、沸石粉、硫酸亚铁、硫酸锰、硫酸钴等等铅、铬、镉含量很低,石粉中20PPM左右,其他的在几个PPM左右钙对铅、铬的测定干扰很大,偏离实际结果很高,矿物质原料中其他金属元素的含量很高,比如石粉中钙含量很高,容易在火焰口形成固体颗粒分裂火焰造成结果偏低很多,请问前处理该怎么办才能测准这三种元素。铅灯稳定性比较差,在0.001-0.010间波动正常吗?该怎么让它稳定下来呢
原子荧光火焰法与原子吸收石墨炉法测定土壤与矿石中微量金的比对研究倪通文 , 范宁云 , 王宁(甘肃省分析测试中心) 摘要: 本文分别采用了原子吸收光谱石墨炉法和原子荧光火焰法对同一土壤与矿石中的微量金进行了检测,比较了两种仪器检测方法的检出限,线性范围,测试速度,测试成本。结果表明,火焰法原子荧光光谱法检出限于原子吸收石墨炉法基本一样,线性范围、分析速度、测试成本都优于原子吸收石墨炉法。关键词:原子吸收石墨炉法;原子荧光火焰法;微量金引言 目前,有关测定化探样品中的微量金及矿石中的常量金文献报道中都是对样品先进行分离富集,再采用质量法、容量法、原子荧光法及原子吸收光谱等方法进行测定。当前在微量金的测试中,仪器分析占有主导地位,而每年进口的分析仪器花费了大量的外汇,国内的仪器比重较小。本文主要对使用了德国耶拿ZEEnit-700P原子吸收光谱仪石墨炉法与国产SK-2003双道原子荧光光谱仪火焰法测定化探样品的微量金及矿石中的常量金两方法进行检出限、精密度、分析速度以及直接耗材比对,以找出两种仪器在测定微量金及常量金的优缺点。1实验部分1.1仪器与试剂SK-2003双道原子荧光光谱仪(北京金索坤技术开发有限公司),高强度空心阴极灯(荧光专用Au 242.8nm 北京曙光明有限公司);耶拿ZEEnit-700P原子吸收光谱仪( 德国耶拿分析仪器股份公司),空心阴极灯(原子吸收专用Au 242.8nm 日本日立公司);马弗炉(KDF- S80日本);盐酸、硝酸、硫脲(均为分析纯);超纯水器(ELGA UVF-MK2英国)18.2МΩ、热源0.005、TOC
各有关单位: 为贯彻《中华人民共和国环境保护法》,保护环境,保障人体健康,提高环境管理水平,规范环境监测工作,我部决定制订《水质 钴的测定 火焰原子吸收分光光度法》等4项国家环境保护标准。目前,标准编制单位已编制完成标准的征求意见稿。根据国家环境保护标准制修订工作管理规定,现将标准征求意见稿和有关材料印送给你们,请研究并提出书面意见,并于2011年1月15日前反馈我部。 联系人:环境保护部科技标准司 谷雪景 通信地址:北京市西直门内南小街115号 邮政编码:100035 联系电话:(010)66556214 传真:(010)66556213 联系人:环境保护部环境标准研究所 黄翠芳 周羽化 通信地址:北京市安外大羊坊8号 邮政编码:100012 联系电话:(010)84934068 传真:(010)84921403 附件:1.征求意见单位名单 2.《水质 钴的测定 火焰原子吸收分光光度法》(征求意见稿) 3.《水质 钴的测定 火焰原子吸收分光光度法》(征求意见稿)编制说明 4.《水质 钴的测定 石墨炉原子吸收分光光度法》(征求意见稿) 5.《水质 钴的测定 石墨炉原子吸收分光光度法》(征求意见稿)编制说明 6.《水质 氨氮的测定 流动注射分析-分光光度法》(征求意见稿) 7.《水质 氨氮的测定 流动注射分析-分光光度法》(征求意见稿)编制说明 8.《水质 总磷的测定 流动注射分析-分光光度法》(征求意见稿) 9.《水质 总磷的测定 流动注射分析-分光光度法》(征求意见稿)编制说明 二○一○年十二月三日
请问火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测矿石中铜铅锌,现在有1mg/ml的铜铅锌标准溶液,怎么配标准曲线溶液 。
引言:铅是可在人和动物体内蓄积的有毒金属。铅中毒以无机铅中毒为多见,主要损害神经系统、消化系统、造血系统和肾脏。近年来,铅接触对内分泌、生殖系统、铅接触女工子代的影响也已引起重视。水中铅主要来自于岩石矿物的溶出和含铅矿山、铅冶炼企业的废水、生产铅化合物的工厂废水。由于水中铅的含量比较低,目前测定水中铅常用的方法有:火焰原子吸收分光光度法、无火焰原子吸收分光光度法、氢化物发生-原子荧光法、双硫腙分光光度法、催化示波极谱法。催化示波极谱法费用较高,而双硫腙分光光度法和火焰原子吸收分光光度法操作比较复杂,本文仅对现在常用的两种方法无火焰原子吸收分光光度法和氢化物发生-原子荧光法的测试做比较,分析两者的优势。1、 实验部分1.1无火焰原子吸收分光光度法实验原理:样品经适当处理后,注入石墨炉原子化器,所含的金属离子在石墨管内经原子化高温蒸发解离为原子蒸气,待测元素的基态原子吸收来自同种元素空心阴极灯发出的共振线,其吸收强度在一定范围内与金属浓度成正比。1.1.1仪器与试剂A3石墨炉原子吸收分光光度计配有一体化自动进样器;铅空心阴极灯(北京有色金属研究总院);铅标准溶液: 1 mg/mL(国家标准物质中心)铅标准使用液:1 μg\mL ;浓硝酸,优级纯:,HF均为优级纯;实验用水均为娃哈哈纯净水。为确保测定结果的准确性,所用玻璃器皿均用10%HNO3浸泡24 h以上,最后用超纯水冲洗晾干后使用。1.1. 2石墨炉测定条件的选择原子吸收分光光度计的工作条件见表1、表2。http://ng1.17img.cn/bbsfiles/images/2012/08/201208200934_384497_2063536_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208200935_384500_2063536_3.jpg1.1. 3实验方法取5~200 mL水样,加入5~10 mL的浓硝酸,在电热板上加热煮沸,蒸发至1~2 mL左右时,取下冷却,用1+99的硝酸定容到至50 mL的容量瓶中,同时制备试剂空白溶液待测。以1+99的硝酸做空白稀释液,由自动进样器将10 ng/mL铅标准使用液稀释成相应的标准系列溶液,按表2的设定工作条件,测定标准溶液和样品溶液吸光度,仪器自动绘制标准曲线并计算出稀释后样品溶液中的铅含量。1.1.4实验结果1.1.4.1标准曲线绘制了铅的标准曲线,铅的回归方程为A=(0.004016+0.00112)/(1+0.00288c),R=0.9998;测定200ng/mL铅标准使用液,铅元素的特征浓度为3.215 ng/mL; 1.1.4.2检出限及线性范围连续测定空白11次,求的空白荧光值的标准偏差,后建立标准曲线。用3倍空白荧光值的标准偏差除以标准曲线斜率即得出仪器的检出限(仪器进样量为1mL):按DL=3S/K(其中S为空白溶液11次测得标准偏差,K为校准曲线斜率)计算。结果求得该方法检出限为0.106ng/mL。按10ml的取样量,其线性范围为0~100μg/L1.1.4.3精密度实验对三个水样连续测定6
麻烦问一下大家用安捷伦火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定元素,采用标准加入法,试剂空白是什么?最后还要进样品,这个样品是什么?
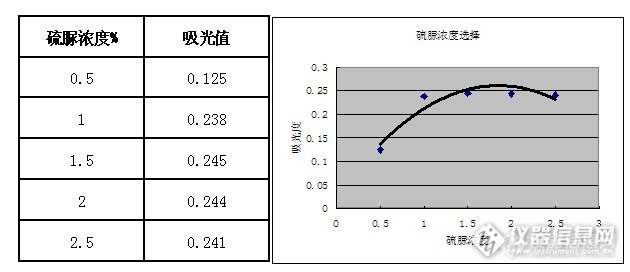
泡塑吸附—火焰原子吸收光谱法测定矿石中金含量【摘 要】本文主要采用泡塑吸附-火焰原子吸收光谱法测定矿石中金含量,对溶液酸 度、吸附时间、解析时间、硫脲浓度等条件进行探索,得出优越的实验条件。【关键词】泡塑吸附 火焰原子吸收光谱法 金 条件选择 金在自然界含量极低,根据最新研究成果,金的地壳丰度值仅为1.1×10-3g /t。随着金矿的普查勘探,对金的分析提出了更高的要求。目前,金的富集方法很多,主要有铅试金法、活性炭吸附法、溶剂萃取法、泡沫塑料吸附法等; 金的测定方法也很多,主要有重量法、氢醌容量法、碘量法、硫代米蚩酮光度法、原子吸收法、等离子体质谱法等。自20 世纪70 年代发现泡沫塑料对金属元素有吸附性以来,由于这一新的富集技术简便易行、成本低廉,因而得到快速发展,并广泛应用于金的富集分离。本文采用王水溶矿-泡沫塑料富集-硫脲解脱-火焰原子吸收法测定金,并在实验的基础上优化了测定条件。采用硫脲解脱,避免了因灼烧引起的空气污染,方法操作简便,干扰离子少,精密度高,测定结果稳定,重现性好。该方法应用于大量地质样品中金的测定,结果令人满意。1、实验部分1.1 仪器与试剂仪器:原子吸收分光光度计(普析,TAS990)、马弗炉、锥形瓶(250ml)、比色管等试剂:盐酸、硝酸、1.0mg/ml金标准储备液1.2 仪器工作条件http://ng1.17img.cn/bbsfiles/images/2013/09/201309051621_462593_2352694_3.jpg1.3 实验方法1.3.1样品制备(详见帖子http://bbs.instrument.com.cn/shtml/20130706/4835569/)1.3.2称取25.0g金矿样于蒸发皿中,放入马弗炉中630度恒温1h(升温到630度需1h,共两小时);(详见帖子http://bbs.instrument.com.cn/shtml/20130711/4845138/)http://ng1.17img.cn/bbsfiles/images/2013/09/201309051627_462596_2352694_3.jpg1.3.3放冷后转入250ml锥形瓶中,用少量水润湿,加入50ml王水(1:1),在电热板上加热使样品溶解,溶液剩余10ml左右取下,冷却,用水冲洗瓶壁,并加水至120ml左右。1.3.4每个锥形瓶中加入两块准备好的海绵,震荡40min,取下。1.3.5每块海绵用水冲洗30次,放入加有25.00ml的1%硫脲的比色管中,水浴20min。http://ng1.17img.cn/bbsfiles/images/2013/09/201309051628_462597_2352694_3.jpg1.3.6尽快取出海绵,放冷后待测。1.4 标准曲线绘制用1.0mg/ml金标准贮备液逐级稀释至10.0ug/ml金标准工作液。移取一定量的金标准工作液,配置一系列金标准工作系列溶液(浓度及移取体积见下表)按照1.3.4-1.3.6步骤处理标准溶液,并绘制标准曲线。http://ng1.17img.cn/bbsfiles/images/2013/09/201309051623_462594_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/09/201309051624_462595_2352694_3.jpg2、结果讨论2.1加入王水酸度的选择移取金标准工作溶液10.0ml5份于250ml锥形瓶中,溶液酸度分别控制在10%,15%,25%,30%,35%,分别加入两块泡塑,于震荡机上震荡40min,取出泡沫塑料,用自来水冲洗干净; 将泡沫塑料放入25 mL 比色管中,加入1.0 %硫脲的溶液25 mL,置于水浴锅中解脱20 min; 取出比色管,捞出泡沫塑料,挤干。待溶液冷却后,以原子吸收光谱法测定。[/size
氢化物发生—无火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法快速测定水中砷、硒 马学俊 于宝坤 艾铭进 李宝昌 绥化铁路卫生防疫站 ( 黑龙江 152072) 本法建立了以流动注射氢化物发生器 , 采用无火焰电加热式原子化器配合[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]光度法 , 快速测定可生成气态氢化物的砷、硒,从而大大提高了分析速度,缩短了检验时间,且水中共存离子对砷硒的测定基本不干扰。所建立的分析方法准确、简便、快速,特别适用于水中砷、硒的测定。 1 实验部分 1.1 仪器及工作条件 AAS9542C 型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光度计 ( 北京苏辉仪器有限公司产品 ) 采用微机作整机控制及数据处理, 高性能砷、硒空心阴极灯, 流动注射氢化物发生器 , 电热式原子化器 ( 北京瀚时制作所研制 ), 仪器的工作参数及测定条件见表 11.2 试剂 ( 所有试剂均为优级纯 , 用 2 次去离子水配制 ) 1.2.1 10%HCI(V/V) 1.2.2 2% 硼氢化钾溶液 2gKBH4,0.4gHaOH 到入塑料瓶中 , 加去离子水至 100ml 溶解 . 1.2.3 砷标准溶液 :1000 μ g/ml 砷标准溶液 ( 国家标准溶液 GSBG 62027 — 90) 用 10%HCI 溶液稀释成 1 μ g/ml 砷标准应用液 . 1.2.4 硒标准溶液 :1000 μ g/ml 硒标准溶液 ( 国家标准溶液 GSBG 62029-90) 用 20%HCI 溶液稀释成 1 μ g/ml 硒标准应用液 . 1.2.5 15% 碘化钾溶液 称取 15g 碘化钾 , 溶于纯水中并稀释至100ml, 贮于棕色瓶内 . 1.3.1 工作曲线 砷标准工作曲线 : 于 50ml 标准系列管中分别加入砷标准应用液0;0.50;1.00;1.50;2.00;2.50ml, 分别为 :0;0.50;1.00;1.50;2.00;2.50ml 砷加纯水至刻度 , 再加入浓 HCI5.0ml 碘化钾溶液, 混匀后放置 15min 后测定,线性相关系数 :r=0.9996.CV 值为 5.7%~1.3(%)。 硒 Se(IV) 标准工作曲线 : 于 50ml 标准系列管中 , 分别加入硒标准应用液 0,0.50,1.00,1.50,2.00,2.50ml, 分别为 :0,0.50,1.00,1.50,2.00,2.50ml 硒 . 加纯水至刻度 , 再加入浓 HCI5.0ml, 混匀后测定。线性相关系数 :r=0.9994.CV 值为 6.0%~0.4(%)。 1.3.2 样品测定 1.3.2.1 砷的测定:取等测水加入 50ml 标准管中至刻度 , 再加浓HCI5.0ml 碘化钾溶液 , 混匀后放置 5min 后测定。 1.3.2.2 硒的测定 : 1.3.2.2.1 Se(IV) 的测定 : 取待测水样加入 50ml 标准管中至刻度 , 再加入浓 HCI5.0ml 和 2.5ml, 混匀后测定 . 1.3.2.2.2 总硒量 Se(IV) 和 Se(VI) 的测定 : 取 25ml 水样、标准溶液和空白溶液边同5.0ml 浓盐酸置于聚四氟乙烯密溶样杯中, 于微小消解炉内2min 、压力 0.5MPa 取出放冷后测定 。 2 结果与讲座 2.1 原子化测试与读数延时时间 砷的原子化温度在 900 ℃ , 硒的原子化温度在 950 ℃时 , 峰高最为稳定,过低或过高, 对原子化吸收都有不同的影响,读数延时时间设计3s, 积分时间 12s 时 , 测试结果的吸光度为高 , 且稳定 。 2.2 酸度的影响 氢化反应在酸性中进行 , 砷的介质为 10% HCL, 硒的介质为20% HCI 为最佳, 样品测试时分别加入 10%或20%的HCI, 与标样测试同步进行 。2.3 氩气流量控制在 150ml/min, 过高使唤信号下降 , 过小使唤峰值降低 。2.4 干扰离子的影响 在实验条件下 , 对20ng/min 砷、硒溶液进行1000倍的 Cd2+ 、 Cr6+ 、 Mn2+,5000 倍的 Fe3+ 、 Mg2+ 、 Zn2+,200 倍的 Ag+ 、 Bi3+ 、 Pb2+,50 倍的 Hg2+ 、 Cu2+ 的干扰试验 , 实验结果均不产生干扰 , 吸光度改变小于 10%, 生活饮用水中, 上壕离子一般低于此范围, 因此无需加入掩蔽剂即可进行测定 . 2.5 方法的灵敏度、限与精密度 本法检出限 (3SB) 为砷:0.3ng/ml, 硒:0.6ng/ml 灵敏度为砷 :0.2ng/ml 0.0044A, 硒 :0.26mg/ml 0.0044A 精密度 CV(%) 砷 :4.5, 硒 :2.2 2.6 准确度 : 准确度以加标回收法测定 , 结果见表 2, 3 2.7 实际样品的分析测定 用本方法测定了含硒 30 μ g/L 和含砷 10 μ g/L 的样品 ( 为家标准溶液 GSBG 62029-90,GSGB62027-90 稀释后的浓度 ) ,测定结果平均值μ g/L 分别为硒 :29.90, 砷 :9.97, 精密度 CV(%) 为硒 :4.5. 结论:本文运用了氢化物发生一无火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]光度法快速测定水中砷、硒的检出限更低 , 精密度更好 , 该方法干扰少 , 操作简单,每个样品只须 20min 即可检测完成 , 特别适用大批量的检测分析 . 参考文献 1 中华人民共和国国家 GB5749-85.1986 2 生活饮用水标准检验方法注释 , 重庆大学出版社 1993.1 月 3 水和废水监测分析方法 , 第三版 . 北京 : 中国环境出版社 1989 (表格见附件)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=3264]相关附件[/url]
今天做原子吸收测定矿物质元素,点火,火焰颜色呈现白色。然后我降低了燃气流量,火焰颜色变回淡蓝色了。为什么之前用同样的燃气流量火焰颜色呈现淡蓝色,而今天就出现了白色火焰呢?是什么原因导致的呢??

火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定微量钾的增感效应作者:罗盛旭 梁振益 黄必勇临床上根据毛发中微量元素的含量,可诊断人的健康状况,为防病治病提供依据,特别是对儿童的健康检查,若采血样不易被接受,以毛发测定同样可取得较好的结果。所以,检验毛发中微量元素含量,在预防治疗卫生学上具有重要意义。常规火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定毛发中微量钾,由于灵敏度较低,线性范围窄,测定可靠性较差[1]。为此,笔者选用三聚磷酸钠为增感剂,建立了一种应用增感效应的火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定毛发中微量钾的新方法,其测定特征质量浓度为0.007mgL-1,线性范围为0~5.0mgL-1。1 实验部分 1.1 主要仪器与试剂 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]AA-100(美国PE公司) 钾空心阴极灯(北京威格拉斯仪器有限公司) 电子天平(日本)。1000mgL-1标准钾溶液(由北京威格拉斯仪器有限公司提供) 三聚磷酸钠、 HNO3、 H2SO4、 H2O2、 (NH4)2C2O4H2O均为分析纯。1.2 样品前处理 将毛发置于烧杯中,加温热中性洗涤剂水溶液,用手揉搓毛发,洗净,弃洗液,用水冲洗多次,直至无洗涤剂残留,淋干后,放在无水乙醇中浸泡2min,捞出,再置于乙醚中浸泡1min,取出,挥净乙醚,置105℃干燥箱中干燥30min,取出,用手术剪刀剪成3~5mm,备用。称取处理后毛发1.000g,置于100mL烧杯中,加4mL浓硝酸,待剧烈反应后,置沸水浴中加热消化1h,滴加10滴双氧水,继续加热,直至出现棕色气体时,再加10滴双氧水,如此反复进行5次,然后加1滴1molL-1硫酸溶液及1mL饱和草酸铵溶液,再加热40min,取下放冷,移于50mL容量瓶中,加水定容至刻度,备用。 1.3 实验方法 准确称取0.5mL质量浓度为0.1gL-1的钾标准溶液于100mL容量瓶中,加入2.8mL浓硝酸、 1.6g三聚磷酸钠,用蒸馏水溶解,再定容至刻度,得到0.5mgL-1的钾标准溶液。如此配成钾系列标准溶液,利用空气-乙炔火焰在选定的仪器操作条件下测定钾的吸光度值,绘制标准校准曲线,据此测定样品中钾含量。1.4 仪器操作条件 本实验测定钾的操作条件为:波长λ=766.5nm 灯电流I=6mA 狭缝宽度slit=0.7nm 燃助比qv(C2H2)∶qv(空气)=1∶3。2结果与讨论2.1 增感剂的选择 由添加增感剂前后吸光度值的增量ΔA,比较了乳化剂-OP、 十二烷基硫酸钠(SDS)、 十二烷基苯磺酸钠(SDBS)、 三聚磷酸钠(STPP)等表面活性剂对钾的增感效应[2,3],结果如图1所示。可见STPP对钾产生最为明显的增感作用。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605261851_19075_1634962_3.jpg[/img]由图1可看出,随着STPP质量浓度增加,ΔA值也增加。进一步实验,得到图2结果。从图2可看出,三聚磷酸钠的质量浓度为16gL-1时,其对钾的增感效应较佳。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605261852_19076_1634962_3.jpg[/img]2.2 空气-乙炔流量比的影响 固定空气流量(6Lmin-1),改变乙炔流量,测定增感效应的变化,得到图3所示结果。由图3可见,随着乙炔流量的增大,三聚磷酸钠对钾的增感程度先增大后减小,出现一极值。因此,qv(空气)∶qv(乙炔)=6∶2,即qv(空气)/qv(乙炔)为3.0时,增感效果最好,此时火焰为稳定的还原性富烧火焰,有利于钾的原子化。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605261852_19078_1634962_3.jpg[/img]2.3 酸介质的影响 比较硝酸、 硫酸和盐酸等酸介质的影响,结果显示,硝酸介质中三聚磷酸钠对钾的增感效应较为明显。改变硝酸介质的酸度,按增感效应法测定1.0mgL-1钾的吸光度值,得到图4结果。由图4可看出,硝酸介质的酸度在0.4molL-1时,增感效果较好。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605261853_19079_1634962_3.jpg[/img]2.4 共存离子的影响 按增感效应FAAS法测定含1.50mgL-1钾的毛发样品试液,共存离子对测定均无干扰。 此时,各共存离子的质量浓度为(mgL-1):Na+(17.2) Ca2+(31.9) Zn2+(4.5) Mg2+(1.6) Pb2+(0.7) Cu2+(0.4) Hg2+(0.08) Ni2+(0.07) Mn2+(0.06) Cd2+(0.03) Co2+(0.01) As3+(3.2) Fe3+(0.5) Al3+(0.3) Cr3+(0.04)。 2.5 校准曲线 配制一系列钾的标准溶液,以增感效应FAAS法测定其吸光度值,得到标准校准曲线,结果见表1。 [img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605261853_19080_1634962_3.jpg[/img]2.6 方法的灵敏度与检出限 由增感效应法与常规法分别测定0.5mgL-1钾标准溶液的吸光度值,参照常规法的有关定义[4],计算2种方法的灵敏度与检出限,结果如表2所示。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605261854_19081_1634962_3.jpg[/img]表2结果说明,增感效应FAAS法较常规FAAS法的测定灵敏度提高10倍,其检出限降低10.5倍。2.7 样品分析 准确移取25mL备用样品溶液于50mL容量瓶中,加入1.4mL浓硝酸、 0.8g三聚磷酸钠,用蒸馏水定容到50mL。利用校准曲线法按实验方法测定样品中的钾,平行测定10次,其RSD为0.85%~2.50%。取上述样品溶液25mL,加入质量含量为0.40~1.40mgL-1的标准钾,测得其回收率在100.3~102.6之间。参考文献:[1]邓勃.我国火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]分析技术的发展[J].分析仪器,2000,(1):1-12。[2]乔元彪,阎卫.增感效应火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定矿石中的微量铬[J].分析试验室,1999,18(3):68-70。[3]冯庆彩,郑斯运.表面活性剂在火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法中增感效应的研究[J].高等学校化学学报,1990,11(8):1043-1045。[4]朱明华.仪器分析[M].北京:高等教育出版社,1995.321-323。
请问大家火焰原子吸收法测定药物中锌、铁含量的方法回收率应达多少?在论坛搜索了一下,有说85%-115%、也有95%-105%等,能不能提供权威的标准名称?谢谢

火焰原子吸收法测定金矿样品经验分享 接触火焰原子吸收法测金已经有7个年头了,期间也遇到过很多问题,从中也不断学习和积累了一些浅见,供大家参考。原子吸收法测试的影响因素很多,有气体方面的,有仪器状态方面的,也有标准溶液方面的,还有样品前处理方面的;在论坛也看到很多新手就以上这些方面发出过求助帖子,下面就这几个方面一一阐述,不足之处请大家批评指正。一、气体方面气体主要有燃烧气和辅助气,燃烧气大多用的是乙炔和笑气,我用过较多的是乙炔;辅助气用的是空气。燃烧气和辅助气之间的各种情形,安老师已经给大家分享了,我就不班门弄斧了。我这里要强调的是,乙炔纯度一定要高纯的,http://ng1.17img.cn/bbsfiles/images/2014/11/201411142321_523163_1657564_3.jpg否则,你看到的火焰颜色不是蓝色的而是桔红色的,http://ng1.17img.cn/bbsfiles/images/2014/11/201411182204_523575_1657564_3.jpg这还不要紧,要命的是,你好不容易画出了工作曲线,没过多久(也许就分析完20个样品),你的QC(质控标准)就偏高很多了,比如说你的QC正常值是5.0,偏差允许范围是2%的话,那你的值范围就是4.9~5.1之间,但是实际上你的QC测得值已经是5.5或者更高了,然后你观测火焰,发现火焰很不稳定,像是有风在吹着它似的;如果你熄火拆下燃烧头,很可能上面已经有黑黑的积炭了。我清楚记得刚接触的时候,以为点着火能测试就行了,其实不然,碰到过连续换了3瓶气都是这样的,怎一个郁闷了得。所以说,乙炔必须得是高纯的,千万别贪便宜,不然,受罪的就是你。再说辅助气,空气压力要达到分析要求,不然可能点不着火的,空气通常是用空气压缩机来获得的,这里也要提醒大家,湿度大的时候要给空气压缩机排排水,空气压缩机不能用有油的哦。二、仪器状态方面仪器状态好不好,测试人员最有发言权也是最了解的,状态好的话,测试起来得心应手,一大批样品很快搞定,结果是准确可靠,那叫一个高效率;状态不好的时候,测试的结果自己心里都没底,几个样品够你折腾大半天的。那可能有人要问了,什么样的状态才叫好?引用国标中的一个判断条件:http://ng1.17img.cn/bbsfiles/images/2014/11/201411132202_522980_1657564_3.png简单的来说:就是工作曲线的线性要好,不说0.9999,至少也是0.99以上吧,其次是QC结果在合理控制范围内,如果标准物质的结果也跟参考值吻合的话那是再好不过了。请看下图工作曲线:http://ng1.17img.cn/bbsfiles/images/2014/11/201411142052_523143_1657564_3.jpg测试数据(删除一些数据,重点看QC)。http://ng1.17img.cn/bbsfiles/images/2014/11/201411141058_523037_1657564_3.bmp那状态不好是什么情况呢?很可能你的工作曲线线性很差,画完曲线测QC样品都显示超出范围或者Wrong(错误)。这时候你需要进行仪器的调节,优化状态或者清洗、清洁仪器光路、燃烧头、雾化器等。看下图数据蓝色部分为重点关注区域:工作曲线:http://ng1.17img.cn/bbsfiles/images/2014/11/201411142100_523147_1657564_3.jpg测试数据(看波动、振幅大小)http://ng1.17img.cn/bbsfiles/images/2014/11/201411141059_523038_1657564_3.bmp还有就是,点火前要确认乙炔和空气的压力是否达到要求的压力,否则要么点不着火(空气压力不够);要么火焰很不稳定,并逐渐变瘦(乙炔压力不够)。三、标准溶液方面说起这个有人会不以为然,有时候你曲线做的很漂亮,QC也很好,但是测试标准物质结果就是偏高或者偏低5%,在排除了前处理、仪器状态、气体方面的情况下,你是不是该怀疑一下你配制的标液是否有问题?你进行过比对或者验证吗?是和上次的还是外购的标准溶液比对?我们有一次,我休假几天,刚好溶液用完了,一个同事帮忙配制的标准溶液系列,测试的时候没来及验证,结果测试结果比往常高了3%(通过标准物质看出来的),最后证明是标准溶液整体配低了才导致结果就相对偏高了。所以说,配制标准溶液也是检验一个分析检测人员的个人技能的一个手段。四、样品前处理如果是严格按照标准方法或者企业内部经过验证的方法来进行处理的话,一般是不会有什么问题的;但是有人员做事不高仔细认真,或者叫不够严谨的话,或许体积就多了(或者少了)那么一点点,这样溶液的浓度也就不一样了,看似很细微的差异,放大就很厉害了。拿我们的金溶液来说吧,通常最后体积是控制在4ml,但是如果你多了0.2ml,那就是5%的偏差了,那你还拿什么来保证你结果偏差在5%以内呢?检测人员在拿到样品溶液的时候最好先确认一下体积是否正常,如果相差太大就有问题了,这样测出来的结果你还觉得可信吗?小结通过以上几个方面的絮叨,或许对你遇到的类似的情况可以提供一些参考和借鉴,快速找到解决问题的方法和途经。以上是个人的经验之谈,不足之处,还请专家和老师补充和完善。
火焰型原子吸收分光光度计测定大米中钾元素时,分别采用湿法消解以及微波消解两种前处理方法,然后还试过用纯水以及2%的硝酸去定容样品前处理液,钾标准液也是用2%硝酸去定容的,用火焰型原子吸收分光光度计测得的钾元素标准曲线达到99.9%以上,但是浓度越高的加标样品反倒测出来的钾含量越低,甚至比无加标的时候还低,不知道问题出在哪里了?
GB/T 15923-2010|镍矿石化学分析方法 火焰原子吸收分光光度法测定镍哪位大侠有下有这个标准的吗?有的话共享一下啊,我的邮箱是:QJY_2008@163.com CANS评审要用,谢谢了
[em0712] 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定地下水中钾钠的试验研究塑。地下水中含有较多的钾, 钠离子, 这是因为它们的盐类有广泛地分布和较大的溶解度 钾离子是植物所必需的营养元索。含钠盐(如碳酸钠或碳酸氢钠)较高的水作为锅炉'用水是不和的。因为这种水加热后,会产生夫量co 而引起泡沫现象。如作为灌溉用水, 对干旱地区会引起土壤盐碱化,直接危害植物生长。因此, 在当今地下水水资源合理开发利用中,首先要了解地下水水质状况, 其中包括钾锕离子含量的分析研究。钾钠离子含量的分析方法有火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法,火焰原子发射光谱怯, 化学法等,其中原子吸收法更有其优越性。在去年河北水利系统对河北省地下水水质状况进行大面积普查工作中, 我们应用火焰厦子吸收法对邯郸, 石家庄, 衡水、邢台四个区域222个测井地下水中钾,铺、镉,镀项目进行了分析试验研究,取得了较为满毒的结果。下面仅对钾. 钠项目的分析试验研究予以介绍。实验部分一、使器1)GFu一201型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计(北京分析仪器厂产)2)钾元素灯(北京有色金属研究总院产)3)钠元素灯(4E京有色金属研究总院产)4)空气压缩机5)钢瓶装乙炔供气二、试耕1)钾标准储备溶液: 称取0.9534[~在150。c烘箱内烘干2小时的基准氯化钾置于250m1烧杯中,以去离子2小时溶解后, 移入50oral容量瓶中,用去离子水稀释至刻度,摇匀,其浓度为1.O00mg/ml。2)铺标准储备溶液: 称取 1.盯1拒在150 c烘箱内烘干2小时的基准氯化蚋置于250ml I烧杯中,以去离子水溶解后, 移入500rnl容量瓶中, 用去离子水稀释至刻度, 播匀,其浓度为1.O00mg/ml。3)钾, 铺标准涟用液: 用钾标准储备液, 以去离子水稀释成浓度为ioo~c/ml和10 g/ml。用钠标准储备液以去离子水稀释成浓度为100~g/mlo4)(1+1)硝酸: 以优级纯浓硝酸与去离子水等津积混合o5)电离缓冲剂:l嘶(m/v)硝酸铯溶液。三, 仪器工作条件经过反复实验,找出仪器艟佳工作条棣.如下表四, 测定步骤1)标j隹系列配钮 1 00mJ容量糯 申维普资讯 http://www.cqvip.com第3期 光谱仪器与分析 4l表1 仪器最佳工作条件鬲哥哥哥注 涮钾时,璐烧头逆时针转25度角分别取适量标准使用液, 并加入4m1(1+1)硝酸,6m1硝酸铯液,用去离子水稀释至刻度, 配成两个标准系列:K(v.Jm1)o o.2.o.4、o.6、O.8、1.o,1.5、2.oNa(p.g/m1)o 1.o、2.o、4.o、8.o 8.o 1o2)测定水样配制:视水样中钾钠离子含量不同,准确移取1—5oml原水样或经过滤后的水样(要经过反复实验确定水样稀释倍数),于1ooml容量瓶中,分别加入4ml(1+1)硝酸,6m1硝酸铯渡,用去离子水稀释至刻度, 播匀。8)开启仪器,选取上述仪器工作条件,用试帮空白液将吸光度值调零,测量标准系列各点吸光度及各水样的吸光度。结果与讨论一, 嗣定结果1)将标准系列各点_浓度值和相对应的吸光度值依次输入pc— I5oo计算机(计算机内先输入计算程序),计算机即打印出标准曲线方程, 相关系数及T检验结果如下:k( g/m1)】【0 0.2 0.4 0. 6 0. 8 1.0 1.5 2.0y 0 0 18 27 36 44 66 88y= 0.331+43,0x R=O.9999 T.=1.751Na (~g/m1)x 0 1.0 2. 0 4. 0 6.0 8.0 t0y 0 7 14 25 35 4By=1.53+ 5.59X R = 0. 999 T .= 2.375注: 当相关系数和T检验不合格时要重新测定。2)根据K,Na的标准曲线方程, 各承样的吸光值和水样舳稀释倍数以及仪器的量程衰减倍数确定备水样的浓度值(计算盛式略)部分结果(m 1)如下邪郸73个铡并水样K最大值为21.B,最小值为0.1BNa最来值为l127,最小值为5.40石家庄48个测井水样K最|大值为 85,最小值为e 2。Na堆大值为l81.O,最小值为4善.8衡承52个测井水样 .K最大值为6.‘g,最小值为o ‘lNa堆大值为876.8,最小值为7$.3邢台51个测井水样K展大值为12.4,最小值为0.sgNa最大值为840.0,最小值为11 23)准确度检查:从四个区域内任取若干个水样傲回收率试验,部分结果如下表;由表二可知;加标回收率钾在96.O ~1o7嘶之间,钠在95.5嘶~1o4嘶之间。基丰符合水质分析之要求.;, 讨谗维普资讯 http://www.cqvip.com 匏。 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定地下水中钾,钠的试验研究裹2 匝 噶试验结果加^ 量 回收量 回收率鼠域名 元 素(pg/mI) {pg/mI) (嘶)鄙 郸 K 1.00 1.02N.a B. 0D 8.韶石京庄 K 1.O0 D.96 目6.0N a B.~-3 7 87 哺.4K 1. 衡 永 D0 1 Dr l●●N a 8.oo 7 64 95. 5K 1. ⋯ 邢自 D0- 1. 4NB 8. 00 8.19 ●∞ 一1)工作条件的选择:仪器的每一项最.佳工作条件都要经过反复l试验来礴定。样品较多时, (如本次222个释品),在测定过霉中要保证仪器前各工作条件不变J要经常检查吸光度零点,要经常用标准液检查或校正,使标准液吸光度值测量前君保持不变,以箍高桦鼯测量的准确度。2)醺度和电离缓冲l剂加入量试i验 经多次试验硝酸酸度在1孵~ 钾0以内耐j其}定结果真宣影响t如测样体积 口mI加^ (1+1)酮晦一12ml测量缚果完全一致,而且做回1发章试验亦符合要隶。本次样品澍老硝酸酸度 0骺(V/V))。电禽缓冲剂加入量应鹦样体积大小和钾蚺含羞来确定。本试验采月i电离缓冲耕韵量为口% (、 V), 经多次回收率试验, 果无显著差异。3)水样稀释倍数的确定:地下水中钾锕离子含量较高,一般都要通过稀释方 测定。因各地下水样所在地理位置不同, 埋探不一,备水棒中钾,钠含量相差甚太, 翁永榉的稀释带来很大困难。初次测定时,首先稀释一倍(其中包括加入硝酸量和也离缓冲剂的量0进行测定。当吸光度谨超出标准血线范围瑚L,视超出量韵太小来确定稀释倍数。个别水桦要经过反复试验,直至吸光度值落在标准曲线范围内。当稀释倍数太大时, 可使用仪器l的 基程衰减捎使其吸光度值降低[~76477~]
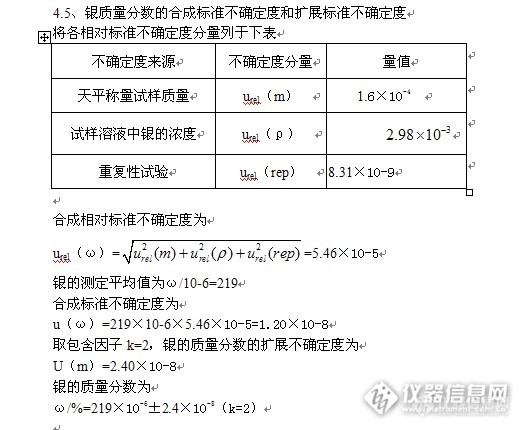
原子吸收光谱法测定矿石中银的测量不确定度评定http://ng1.17img.cn/bbsfiles/images/2013/07/201307151003_451288_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151004_451289_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151004_451290_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151005_451291_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151005_451292_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151005_451293_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151007_451294_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151007_451295_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307151008_451296_2352694_3.jpg
纠结了很久的原子吸收火焰测定水中钾的问题:国标中注明用酸进行标准物质的定容,而实际质控的操作中,盲码要求用纯水定容,两者互相打架的问题解决了,今年新来的一批盲码说明略作改正,既可以使用纯水也可以使用酸类定容,对我们是利好消息。
求助此标准,GB/T 15923-1995|镍矿石化学分析方法 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法测定镍量谢谢各位
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测锌的一些故事:[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定锌,由于其灵敏度极高而广受青睐,在火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]初始阶段就被广泛应用。由于灵敏度高,测定波长又较短,并且没有灵敏度相当的其他分析线又给它的测定带来了麻烦。1.由于其测定波长(213.9nm)较短,化学剂量的空气乙炔火焰通常要吸收其40%左右的辐射能量,火焰的抖动(这个吸收也同时变化)造成较高的闪变噪声,成为测定信噪比好坏的重要因素。火焰中气体成分的变化对锌谱线的吸收也会变化,在没有背景校正的情况下,基线会发生漂移,使测定的检出限变坏。经常会发现即便吸喷蒸馏水也能读到吸光度读数,尽管读数很小,在浓度较高的测定时不易觉察,但是在检出极限附近,这个值就很可观了。也就是说,在低浓度测定时往往需要背景校正!良好的背景校正系统能消除火焰燃气流量变化造成的漂移,也能有益于改善火焰的闪变噪声带来的影响。这也就是我在特邀论坛时谈到火焰塞曼能改善短波元素测定检出限的原因,马怡载老先生在一次海水中锌的测定仲裁中专门找我用塞曼火焰直接进样进行了高精度的测定。信不信由你:早期我国生产的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]仪器需要做六个元素的检出限(Zn,Cu,Mg,Ni,K,还有什么元素我都忘了,那是1976年上海光学仪器研究所主持制订的国内第一个[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]仪器鉴定标准,我是制订者之一,那个标准制订会上大家都大吵大闹了半个月。)测定锌的检出限通常是比较费劲的,因为除了仪器光学系统电子系统原子化系统外,空压机必须得好!一次我在鉴定一台新型号的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url]时,用了一台带储气桶的噪声极大的空压机,只好把它放在外面气房里。做锌了,把量程放到最大(那时没有计算机,全是模拟电路,模拟对数变换,用记录器观察吸光度),点火以后调零,基线一会儿向上跑(吸光度增加),过一会儿又往下跑(吸光度降低)。好象很有规律,我们傻了,灯已预热很久,电路有问题?电路设计制作得很好的铜基线能半小时只飘移0.002吸光度!突然,我们发现基线的漂移和空压机启动与停止几乎是同步的!我的好友邓宜成先生(他可是一位[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]专家)乒乒乓乓从放空压机的气房到仪器房跑来跑去验证着这一现象,终于确定:由于空压机外的稳压阀不够精密,在空压机启动(通常从低限启动到高限停止)到停下来缓慢放气的过程中,输出气压微弱的变化,造成了空气流量的变化,继而使基线漂移!后来通过一些改进,仪器通过了鉴定,经验也就留下了。2.锌的光谱干扰:很可怜的锌,主要分析线只有213.9nm(213.856nm)一条,却有一个几乎没有办法克服的光谱干扰线,那就是铁的吸收线(213.859nm),两者之差仅为0.003nm。利用现有的各种背景校正方法都没有办法克服其影响。如果样品溶液中含有2000mg/L的铁(当然这很高)不用背景校正可能产生0.11吸光度,利用氘灯背景校正可能还会得到0.02以上的吸光度,用S-H法背景校正则有0.012左右。如果用塞曼发那就更糟了,所得吸光度为负值,而且很高,因此,做锌的分析应该注意样品的组分中铁的含量。在石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中,这种干扰是很容易对付的,因为锌的原子化温度比铁低很多,调整石墨炉加热参数可以轻而易举地令两者分开蒸发。另外,光谱干扰总归只是邻近线,所以如同曲线变弯继而变平的原理,当干扰物达到一定浓度后干扰产生的吸光度便不会再增加,这就使得钢铁样品中的锌的分析变得容易。也就是说:如果样品中产生光谱干扰的基体浓度很大也很一致,并且对待测元素没有化学干扰或干扰程度比较恒定,仍可使用标准加入法!(当然石墨炉可就不行了,因为蒸发速度的不可控这种光谱干扰也很难一致)信不信由你:儿童发锌测定在一段时间内很热门,有的单位靠这个还发了财,当然如何“发财”的并不在我们的议题中。我单位由于级别高仪器好,承担了北京儿童医院的发锌测定任务,医院负责采样(在儿童头上不同部位剪下头发,清洗过后送到我单位)我单位负责处理测定。多少批结果报上去了,“生意”真好!我们用塞曼[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定速度极快!有一天,医院找上来了,说:“你们的分析结果有问题,肯定!与我们观察的临床效果不同。”我大惊失色。既然要查,那就拿样品重新处理再测。所测结果医院又认可了!两次结果差异很大!问题在哪里呢?多次摸索,发现: 锌极易受污染,绑在容量瓶上的橡皮筋里含锌很高,不小心怎么跑到瓶子里去了,造成污染。污染这个现象是很不能重复的,有时简直是不可想象的(石墨炉更是如此,以后我们还会有故事的)或者说很难理喻的! 第二个原因是:我们的操作人员马虎,电热板上处理的样品用生了锈的大镊子去夹!镊子表面上的铁锈掉入样品,造成了光谱干扰,大量的铁在塞曼火焰法中产生了负干扰。明明很正常聪明的孩子,测定结果是“严重缺锌”!!! 找出原因后大家更注意分析技术了,免不了还感叹这锌的临床效果还真准!同时,真得注意:客户骂你时,还得放下架子好好自我检查的呀!
由于贵金属矿石资源的日趋紧张,贫、散、杂的问题已得到广泛重视。请大家集思广益,谈谈如何才能利用火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定低含量铂。相互交流以下经验、共同提高。[em0803]
以下是我们参与《土壤环境监测分析方法》(魏复盛老师为主编)的内容,版权归该书所有;同时在我们的实验过程和编写中,得到南京站、浙江站、湖南站、广西站等同行、论坛网友、普析和耶拿工程师的帮助、支持,在此表示万分感谢。 铁(Fe)的原子序数是26,位于化学元素周期表中第四周期第Ⅷ族,主要化合价为0、+3、+2,熔点和沸点分别为1535 ℃、2750 ℃,第一电离能和第二电离能分别为762.5 kJ/mol、1561.9 kJ/mol。 铁是地壳含量第二高的金属元素,在土壤中分布差异较大(1 %~15 %)。它是人体和动植物必需的微量元素,参与一系列吸收代谢和生理功能;土壤中有效铁的含量会受多种因素的影响如土壤pH、有机质、氧化还原电位以及养分之间的相互作用等,容易导致缺乏现象。 土壤中铁的测定主要有容量法(重铬酸钾法)、比色法(邻菲罗啉分光光度法)、仪器法(火焰原子吸收法(FLAAS)、电感耦合等离子体-发射光谱法(ICP-OES)、X-射线荧光法(XRF))。多元素同时分析优先选择XRF法、ICP-OES法;单元素分析可以选择容量法、比色法、FLAAS法,FLAAS相比其它两种分析分析方法具有操作方便、设备较贵的特点。1. 方法原理 样品经消解后的溶液直接喷入空气-乙炔的富燃性火焰中。溶液中所含被测元素离子在火焰高温下原子化,形成的基态原子对同种元素空心阴极灯发射的特征谱线产生选择性吸收,其吸光强度在一定范围内与待测元素含量成正比。2. 适用范围本方法适用于土壤和沉积物中总铁的测定。称取0.2 g试样消解定容至50.0 mL时,本方法的检出限为25 mg/kg,测定下限为100 mg/kg。(未考虑样品稀释倍数(25~100) )3. 干扰及消除特征谱线302.107 nm附近存在铬(302.135 nm)、钌(302.088 nm)等谱线重叠。样品中铁含量远比它们高,采用较小的狭缝,可以忽略其影响。4. 仪器和设备4.1 仪器设备火焰原子吸收分光光度计、空心阴极灯、数控平底石墨电热板(最大加热温度不小于350 ℃)、全自动消解仪、微波消解仪、聚四氟乙烯坩埚等。4.2 仪器条件火焰原子吸收分光光度计参考条件见表1。各种型号的仪器,测定条件不尽相同,应根据仪器说明书选择合适条件。表1 仪器参考测量条件特征谱线/nm火焰类型灯电流/mA通带宽度/nm302.1 氧化型(蓝色)4.0 0.2 5. 试剂除非另有说明,该方法均使用符合国家标准的优级纯化学试剂,实验用水为新制备的二次去离子水。5.1 硝酸(HNO3):ρ =1.42 g/mL。5.2 盐酸(HCl):ρ =1.19 g/mL。5.3 氢氟酸(HF):ρ =1.12 g/mL。5.4 高氯酸(HClO4):ρ =1.68 g/mL。5.5 稀释液,(1+99)硝酸溶液:用(5.1)配制。 5.6 盐酸溶液,1+1:用(5.2)配制。5.7 铁标准贮备液,ρ =1.000 mg/mL:准确称取1.000 g光谱纯金属铁(Fe),用50 mL盐酸溶液(5.6)溶解后转移入1000 mL容量瓶中,用稀释液(5.5)定容至标线,摇匀;亦可使用有证标准物质溶液。5.8 铁标准使用液,ρ =10.0 mg/L:移取铁标准贮备液(5.7)1.00 mL于100 ml容量瓶中,用稀释液(5.5)定容至标线,摇匀。6. 步骤6.1 样品采集与制备 按照HJ/166、GB17378.3的相关规定分别进行土壤、沉积物样品的采集、制备。6.2 试样制备6.2.1 电热法数控平底石墨炉电热板的土壤和沉积物的全消解过程见表2。表2 电热板消解过程步 骤过 程 描 述备 注1称取样品:0.195~0.205 g于干燥的坩埚底部。精确至0.0001 g。2加酸浸溶:加入10.0 mL盐酸于样品坩埚,轻摇后放置过夜。含三个全程序空白。3低温消解:75~80 ℃下消解至剩余液体小于3.0 mL后冷却‚。2.0~3.0 h,显示温度200 ℃。4加混合酸:依次加入5.0 mL硝酸、5.0 ml氢氟酸、3.0 mL高氯酸。 5中温消解:115~120 ℃下加盖消解。1.5 h,显示温度270 ℃。6开盖飞硅:温度不变,每隔5.0 min轻摇飞硅至冒浓厚白烟。1.0~1.5ƒ h,显示温度270 ℃。7消解黑炭:温度不变,加盖使坩埚壁黑色有机物完全分解。1.0~1.5ƒ h,显示温度270 ℃。8赶酸过程:温度不变,开盖赶酸至白烟冒尽、消解液为粘稠状。1.0 h,显示温度270 ℃。9转移定容:坩埚稍冷,实验用水溶解残渣、转移、定容至50.0 mL比色管。 注:盐酸、硝酸、氢氟酸、高氯酸依次为试剂5.2、5.1、5.3、5.4,表3~4同此。‚消解达到要求后应冷却。ƒ该过程可以取1.5 h以求消解过程同步。6.2.2 全自动法全自动消解仪的土壤和沉积物的全消解过程见表3。表3 全自动消解仪消解过程步骤过 程 描 述步骤过 程 描 述步骤过 程 描 述1加入0.5 mL实验用水10加入6.0 mL氢氟酸19170 ℃加热15 min250%高度20%强度下震摇
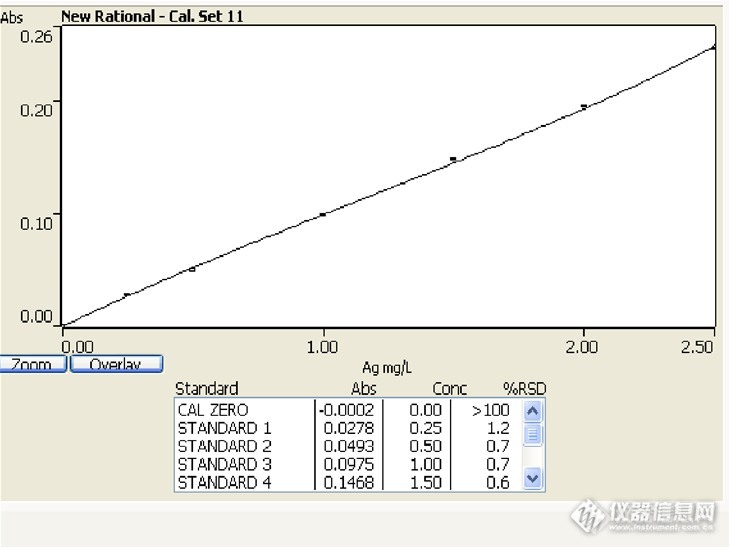
CMA盲样考核之--锌精矿中银含量的测定方法摘要:试料 以 盐酸、硝酸溶解。在稀盐酸介质中,于原子吸收光谱仪波长328.1 n m处,用空气一乙炔火焰,测量银的吸光度。1.试剂1.1 盐酸ρ1.19 g /mL),优级纯1.2 硝酸ρ1. 42 g/mL),优级纯。1.3 盐酸(1+1).1.4 硝酸(1+1).1.5 1000μg/ml银标准贮存溶液1.6 50μg/ml银标准溶液:移取5.0 0mL银标准贮存溶液(3.5 )于100m L容量瓶中,加人4m L硝酸(3.4 ),用不含氯离子的水稀释至刻度,混匀。2.仪器设备2.1 原子吸收分光谱仪,附银空心阴极灯;http://ng1.17img.cn/bbsfiles/images/2013/12/201312312046_486136_1657564_3.jpg2.2 电热板;http://ng1.17img.cn/bbsfiles/images/2013/12/201312312048_486137_1657564_3.jpg2.3 电子天平;http://ng1.17img.cn/bbsfiles/images/2013/12/201312312048_486138_1657564_3.jpg2.4 烧杯2.5 容量瓶3.测定3.1 称量0.5g样品置于250mL烧杯中,以少许水润湿摇散,加15 ml盐酸(3.1),加热溶解,低温蒸发溶液体积至3-5 mL,加入5ml硝酸(3.2),继续加热蒸至近干,取下,稍冷,按表1加人盐酸(3.3),用水吹洗表皿及杯壁,加热煮沸溶解盐类,取下,冷至室温,按表1移人容量瓶中,用水稀释至刻度,混匀。干过滤部分溶液。3.2 仪器参数设置http://ng1.17img.cn/bbsfiles/images/2013/12/201312312053_486140_1657564_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/12/201312312057_486141_1657564_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/12/201312312058_486142_1657564_3.png3.3 使用空气一乙炔火焰,于原子吸收光谱仪波长328.1nm处(银含量小于50 g/t时采用氖灯扣除背景),以水调零测量试液的吸光度,减去随同试料的空白溶液的吸光度,从工作曲线上查得相应的银浓度。3.4工作曲线的绘制3.4.1移取0,0.50,1.00,2.00,3.00,4.00,5.00 mL 50μg/ml银标准溶液分别于至一组100 mL容量瓶中。加20 mL盐酸(3.3),用水稀释至刻度,混匀。3.4.2在与试液测定相同条件下,测量标准溶液的吸光度。以银浓度为横坐标,吸光度(减去“零”浓度溶液的吸光度)为纵坐标,绘制工作曲线。http://ng1.17img.cn/bbsfiles/images/2013/12/201312312101_486143_1657564_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/12/201312312109_486144_1657564_3.jpg4结果计算按式(1)计算银含量:Ag(g/t)_=c·V/m ........................⋯⋯(1)式中:c— 自工作曲线上查得的银浓度,µg/mL;V—试液总体积,mL;m—试 料的质量,g由于测试过程中体积和重量已经输入系统,盲样测试结果为24g/t。







 我要推广仪器
我要推广仪器
 下载APP
下载APP