文献解读丨大鼠血浆中9种三七皂苷绝对定量的UFLC-MS/MS分析方法的建立和验证:三七提取物药代动力学研究的应用
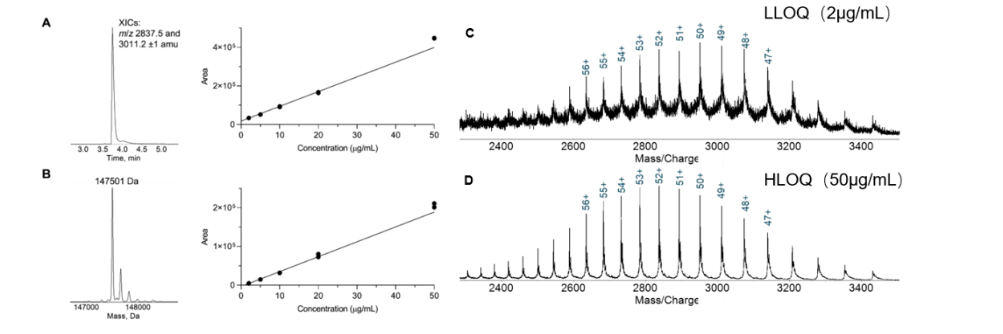
本文由中国药科大学天然药物国家重点实验室药物代谢与药代动力学重点实验室所作,发表在Journal of Chromatography B (2015)46-53。三七皂苷是中药三七的主要活性成分,具有抗氧化、抗高血糖、抗肥胖等多种生物活性。然而,由于三七皂苷在体内浓度低、成分复杂,其药代动力学评价仍然是一项艰巨的任务。 本研究建立了一种基于超快速液相色谱-串联质谱(UFLC-MS/MS)的大鼠血浆中三七皂苷含量快速、灵敏的定量分析方法。三七皂苷R1、Rg3、Rd、Rg2、Rb2、Rf、Rg1、Rb1和Re经正丁醇液-液萃取,在ODS C18柱(5 mm× 50 mm × 2.1 mm)上分离,采用二元梯度洗脱,以负离子模式同时监测,所有化合物均在9 min内进行分析。多反应监测(MRM)方法如下:R1 (m/z 967.7→637.4)、Rg3 (m/z 819.6→621.4)、Rd (m/z 819.6→783.5)、Rg2 (m/z 819.6→475.4)、Rb2 (m/z 1113.4→783.4)、Rf (m/z 835.6→475.4)、Rb1 (m/z 1143.7→945.6)、Re (m/z 981.6→637.4)、内标(地高辛,m/z 815.5→779.4)。验证参数(线性、灵敏度、日内和日间的精密度和准确度、回收率和基质效应)均在可接受范围内,生物提取物在整个储存和制备过程中稳定。将UFLC-MS/MS方法应用于三七提取物在大鼠体内的药代动力学研究,进一步验证该方法的有效性,并利用Winolin软件计算了药代动力学参数。 因此,该方法简便、可靠、准确、精密,可用于各种三七皂苷和其他中药皂苷的药代动力学研究。 使用仪器:岛津LCMS-8050 图1 三七皂苷R1、Rg3、Rd、Rg2、Rb2、Rf、Rg1、Rb1、Re和地高辛(内标)的结构 图2 三七皂苷R1、Rg3、Rd、Rg2、Rb2、Rf、Rg1、Rb1和Re的线性曲线。 图3 空白大鼠血浆和添加R1、Rg3、Rd、Rg2、Rb2、Rf、Rg1、Rb1、Re和IS的大鼠血浆的典型MRM色谱。(A)空白大鼠血浆MRM色谱,(B)添加三七皂苷(50.0 ng/mL)和IS的空白血浆的MRM色谱,(C)大鼠灌胃三七提取物(1.0 g/kg)后2 h大鼠血浆的MRM色谱。 三七是一种在世界范围内广泛使用的植物药,有必要建立一种可靠、灵敏、高通量的方法来测定生物样品中的多种三七皂苷。本文以9种三七皂苷(R1、Rg3、Rd、Rg2、Rb2、Rf、Rg1、Rb1和Re)为研究对象,构建了一个功能强大的中药皂苷药动学分析技术平台。该方法色谱运行时间较短,LLOQs较低,可快速、灵敏地测定大鼠血浆中三七皂苷的含量。此外,该方法专属性强、结果准确、重现性好,已成功应用于大鼠灌胃三七提取物后三七总皂苷的临床前药代动力学研究。更重要的是,目前开发的方法稍加修改后,即可用于其他草药皂苷的药代动力学研究。 文献题目《Development and validation of an UFLC-MS/MS assay for the absolute quantitation of nine notoginsenosides in rat plasma: Application to the pharmacokinetic study of Panax Notoginseng Extract》 使用仪器岛津LCMS-8050 作者Lijun Zhou a, Rong Xinga,b, Lin Xiea,Tai Raoa, Qian Wanga, Wei Yea, Hanxu Fua,Jingcheng Xiaoa,b,Yuhao Shaoa,b, Dian Kanga,b, Guangji Wanga, Yan Lianga a. Key Lab of ug Metabolism hamon y booy of Atul Me Pamaeu Univ. Tongaxang24 Nanjing 210009. Chinab. Department of Pharmacy. The First ffiliated Hospital of Bengbu Medical College, Bengbu Anhui. China























 我要推广仪器
我要推广仪器
 下载APP
下载APP