径向色谱中,圆柱形色谱柱和扇形色谱柱性能上有什么差别?
什么时候使用径向?什么时候使用横向?径向测试与横向测试结果相差很大吗?请各位帮忙解答一下,谢谢!
就普及率来说,轴向矩管的ICP用户比较多,我到现在也还没接触过径向矩管的ICP,那径向矩管的ICP一般应用于什么领域?适用于哪类样品的分析?有何优缺点?
做铝合金分析cu,mg,fe,si,mn,cr,v,ti,zn等,用轴向观测还是用径向观测好呢?
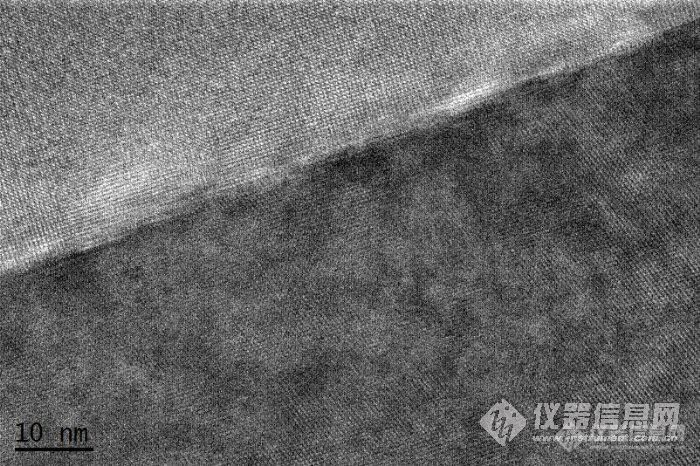
下图为某种辉石矿物出溶结构的衍射谱和高分辨,在衍射谱中出现了众多的卫星斑点,这应该是二次衍射吧,但是感觉斑点不是正规的圆形,存在中间镜像散,但是我来回消中间镜像散,好像难以无法消除,一般各位消除中间镜像散都是如何操作的,是否是IL STIG+ DEF进行操作,那么IL STIG +Shift的作用又是什么?虽然中间镜存在像散,但是好像高分辨还算清楚,请问中间镜像散对高分辨有何影响?http://ng1.17img.cn/bbsfiles/images/2011/07/201107070841_303608_1606080_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/07/201107070843_303610_1606080_3.jpg
单向二次展开薄层色谱法单向二次展开薄层色谱法检识挥发油中各成分时,为什么第一次展开所用的展开剂极性最好大于第二次展开所用的展开剂的极性?单向二次展开薄层色谱法有什么优点?
如题,轴向观测就是水平观测,径向观测就是垂直观测吗?它们有个啥优缺点?谢谢给为指教
面心立方晶格,直到第2近邻的径向分布函数表达。请高手解答谢谢!
谁能介绍以下薄层色谱展开机理和流动相选择[em06] 。谢谢了[em44]

我想请教一个径向分布函数的问题,请大家帮忙。http://ng1.17img.cn/bbsfiles/images/2012/10/201210312300_400569_2629485_3.jpg
请问径向加压柱是什么柱子啊?有什么用途吗?
用途:测试黑色橡胶辊粗糙度,直径5~15mm,粗糙度在零点几个微米到2微米之间。要求:径向运动测试(不是轴向),价格20万以下。
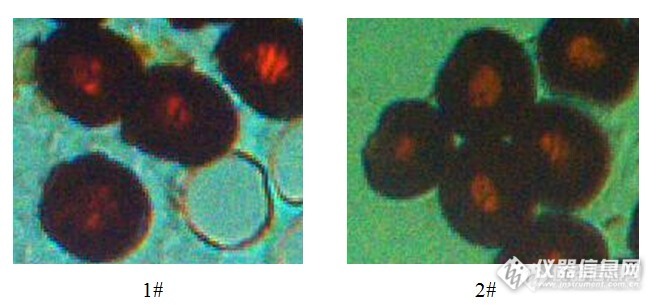
在预氧化过程中,很多研究者认为环境中的氧很难进入到纤维内部是一种物理阻隔。但是从化学角度看,环化反应是氧化反应的前提条件,PAN纤维中生成的环化结构是发生氧化反应的基础,且从物理方面来看氧气分子是极小的,由此推测纤维表层阻碍氧进入芯部是一种化学阻隔,而非物理阻隔。选取两种不同的样品,为了使两种纤维样品的烯胺结构含量不同,将PAN原丝进行预处理,由于在惰性气氛热处理时PAN纤维中烯胺结构含量在从210℃开始迅速增加,1#和2#样品预处理条件分别为在惰性气氛下190℃和230℃热处理12h,再将预处理过的PAN纤维进行相同条件的空气气氛下的热处理,温度为230℃,时间为1h。将两种纤维样品进行核磁测试,可以得到图1。将得到的核磁谱图进行分峰处理,可得出,两种样品核磁谱图中特征峰的相对含量。http://ng1.17img.cn/bbsfiles/images/2015/09/201509241759_567715_3043450_3.jpg图11#和2#样品的核磁谱图表1 1#和2#样品中155ppm和176ppm处特征峰相对含量 1# 2# W155/% 15.48 19.05 W176/% 3.91 4.87 表1中列出了1#和2#样品中代表烯胺结构的155ppm处特征峰的相对含量分别为15.48%和19.05%,代表氧化反应程度的176ppm出特征峰相对含量分别为3.91%和4.87%。1#样品中烯胺结构含量相对较低,相应的其氧化程度也较2#样品低。将两种样品进行包埋,利用切片机进行切片,并在光学显微镜下观察两种样品径向结构的差异。图2为两种样品的径向结构照片。http://ng1.17img.cn/bbsfiles/images/2015/09/201509241759_567716_3043450_3.jpg图2 1#和2#样品的径向结构照片对比两个样品的径向结构照片,可以发现两种纤维都出现了芯部预氧化程度较低的情况,1#纤维的径向结构较均匀,2#纤维出现明显的“皮”“芯”分层现象。这与前面的推测相吻合,由此从化学结构角度提出预氧化过程中PAN纤维径向结构的形成机理。预氧化过程中,氰基发生反应形成亚胺结构的同时,亚胺结构向烯胺结构转变,且在氧气的促进作用下转变的越多;由于与其他结构相比烯胺结构容易被氧化,纤维表层的烯胺结构不断与氧发生反应,导致扩散到纤维内部的氧较少,从而形成内部预氧化程度较低的不均匀的径向结构。
我使用的是PE的5300DV,检测Ca/Mg/Cu/Mn/Fe/Zn/Cr,其中Ca/Mg的标准浓度最大值是200ppm,其他最大值大概是3 - 20ppm,以前一直是使用径向Radial检测器,但是Cu/Zn/Cr的信号值就会很小,大概只有2000左右,测出来的结果就会和标准值有较大差距,约有5 - 10%。后面把方法修改,同时使用径向Radial和轴向Axial检测器,其中Ca/Mg就用径向测,其他元素就用轴向测,提高灵敏度,结果相对合理。但仪器用了大概4个月左右开始出现问题,XY轴切换马达出现故障,使检测器回不到设定位置,只有重新恢复出厂设置才可以。但相同问题很快又会出现,工程师说是切换马达需要更换了,但一个要4w啊,而且还不能保证使用多久。我想请教一下各位大虾,有谁在检测方法中会同时设置X轴和Y轴检测器一起检测的,使用过程中有没有出现问题?
晶体结构中的面心立方,直到第2近邻的径向分布函数表达。不知道晶体的如何表达,请高手解答!谢谢!
扫谱的次数一般为四的倍数,是为了防镜像。那这个镜像是如何产生的?为何扫描次数为四的倍数就没有镜像而不是四的倍数就会有镜像呢?
求有关光谱类的外文文献网址或者镜像
中药材薄层色谱展开系统之浅见 工作几年以来,主要运用薄层色谱分离方法对中药材成分进行分析。薄层色谱直观的色彩图像表达方式是一大特点,通过各种展开剂的灵活运用可将中药材分成不同极性组分进行分离,充分展现出层析分离的意义。根据这几年的摸爬滚打,反复实践,总结出一点展开系统的经验。由于接触的药材有限,有些仍不够深入,因此观点可能有些片面,鄙薄之处望多多包涵!说明:以下使用的板为硅胶正相薄层板 1 黄酮类经典展开剂:甲苯-乙酸乙酯-甲酸(5:4:1),该系统还可用于香豆素类,三萜类成分分离,比例应作适当调整。 2 正己烷-乙酸乙酯或环己烷-乙酸乙酯系统,可分离极性相对较小的系统,如单萜类,木脂素类成分。可适当加甲酸,因己烷系统易使斑点扩散,甲酸对酸性成分分离有帮助。 3 乙酸乙酯-甲酸-冰醋酸-水(15:1:1:2)是甘草的展开系统,但其适用性很广,可用于中等极性的成分如绿原酸,DCQ,黄芩苷等。人参系统氯仿-乙酸乙酯-甲醇-水(15:40:22:10)(冰箱分层取上层溶液)也是一个不错的分离中等极性的展开剂。 4 若改变展开系统对分离帮助不大时,可考虑用变性板,如0.5%或1%NaOH溶液,0.1M NaH2PO4,0.1M Na2HPO4等。变性板还可调整斑点的Rf值,可将其减低。 5 氯仿是药典和文献广泛使用的展开溶剂,由于毒性较大,现在正逐渐用其他溶剂替代。有氯仿的两相系统易产生边缘效应,这与其沸点较低易挥发有关,若在其中加入些许酸或碱对分离可能产生意想不到的结果,特别是中药复方制剂时排除阴性干扰时,用量视情况而定。 总之,展开系统的千变万化使薄层色谱有了更多可操作可摸索的空间,因此有了更为丰富多彩的色谱图像,若能结合其它色谱得知化学成分的一一归属关系就更加深刻了。
根据本人的几年薄层层析经验,参考药典等国家药品标准和有关文献,将 2000版药典一部里部分有代表性的对照品的薄层层实例按展开剂极性排序,并对其规律做一些分析。以下的分析和介绍是总体描述性的,目的是快速、简便地选择展开剂。如果想了解展开剂选择的各种理论,请参考其他专著。 选择展开剂,要依据溶剂极性和他们的混溶性,溶剂对被分析物的溶解性,以及被分析物的结构。这里只讨论药典里通常使用的以硅胶为固定相主体的正相薄层,也不考虑板的活性。 列出溶剂极性参数表,方便以下比较展开剂。环已烷 :-0.2、石油醚(Ⅰ类,30~60℃)、石油醚(Ⅱ类,60~90℃)、正已烷:0.0、甲苯:2.4、二甲苯:2.5、苯:2.7、二氯甲烷:3.1、异丙醇:3.9、正丁醇:3.9、四氢呋喃:4.0、氯仿:4.1、乙醇:4.3、乙酸乙酯:4.4、甲醇:5.1、丙酮:5.1、乙腈:5.8、乙酸:6.0、水:10.2 [1] 。 关于溶剂混溶性,一般根据相似相溶原则,需要注意,极性相差大的不混溶,比如正己烷与甲醇。多元展开剂,主体的两种溶剂不能混溶,就需要通过第三种溶剂来调和。比如:石油醚、正庚烷、正已烷、戊烷、环已烷和甲醇、水之类的。 一般正相色谱,固定相为极性,被分析物质的极性越大,需要极性更大的展开剂。 了解被分析物的极性可以通过分析其结构获得,很难获得它的极性指数。物质分子化学结构中,通常由较极性部分和非极性部分两部分。
ICP-OES的轴向观测与径向观测对其分析性能有何影响?什么时候适合用轴向观测,什么时候适合用径向观测?

我想请教一个径向分布函数的问题,请大家帮忙。http://ng1.17img.cn/bbsfiles/images/2012/10/201210312258_400568_2629485_3.jpg
摩擦色牢度测试时径向和纬向结果相差1级以上的情况有吗?一般是什么原因引起的?
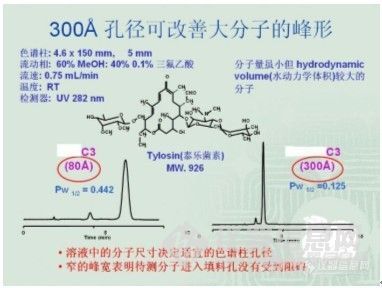
液相色谱方法开发的一般规则 高效液相色谱方法的开发是一个繁复的过程,但不管再繁复,也有其规律可寻。方法开发过程中,一个总的原则是:先找到目标化合物的峰,然后调整峰形,再是进一步完善。1. 找到目标化合物的峰:要找到目标化合物的峰,我们该如何开展工作,先举一个例子,下面是分析氟康唑氯化钠注射液的一个谱图:http://ng1.17img.cn/bbsfiles/images/2010/07/201007081805_229421_1896702_3.jpg谱图上的内容主要包括:色谱柱、波长、流动相、温度、流速和进样量这几项。这意味着如果这几个色谱条件都确定下来了就可以基本认为这个方法已经开发成功,所以开发方法时我们可以通过逐一的考查这几个色谱条件来进行。 考查色谱条件是需要通过在色谱仪上进样来进行的,这就需要我们首先确立一个初始条件,即回答从何开始的问题。在回答好“从何开始”这个问题之前,我们先要了解我们的被分析物,就像一场战争,首先要知道自己的敌人是谁一样,我们要了解它的物理、化学性质,特别是化学结构式非常关键。所以,色谱方法开发工作可以通过如下的步骤来展开:1)收集资料:对它的分子量、结构式,以及在水、甲醇、乙腈、四氢呋喃、正己烷和异丙醇中的溶解度有一个初步的了解。对分子量的了解在选择色谱柱的过程中是非常重要的,因为色谱柱填料的孔径对化合物的分离具有重要的影响,例子如图:http://ng1.17img.cn/bbsfiles/images/2010/07/201007081807_229422_1896702_3.jpg 由此可以看出填料孔径对分离度和峰形是有一定影响的,120A的色谱柱通常适用的范围为分子量10000的,如果分子量太大,在填料为120A孔径的柱子上分离度会比较差,因为样品分子在色谱柱上有较好的保留是由于可以进入到填料的微孔里面,与键合在表面上的C18长链相互作用,通常孔径直径需要大于分子直径的3倍以上才不会对分析造成影响。因此一般分子量10000一下的化合物建议用120A的柱子来分析,分子量大于1W小于20W的用300A的来分析,分子量大于20W的就要用凝胶柱了。 结构式对于分子极性大小的预测以及后续调整峰形时具有非常重要的作用,如-COOH、-NH2、-NHR、-NR2、-OH等都是极性基团,而苯环、己环、-CH=、-CH2-CH3等都是非极性基团,根据经验大致对其极性做一下判断,估计一下可能在C18上(用得最多,我们最熟悉)的保留性能如何,再结合目标化合物在上述所说的几种溶剂中的溶解度状况,对方法开发时可能用正相柱还是反相柱来作一个粗略的判断,以及方法开发完成后的认证有作用。2)选择色谱柱 I)填料孔径的选择:根据所查资料获得的化合物分子量信息来确认色谱柱填料的孔径; II)填料键合相(键合相是指C18、C8、苯基柱等)的选择:在没能查到做过该样品的相关资料之前,或者并不了解其极性之前,通常最好选择C18柱作为初始的色谱条件。因为C18柱是我们用得最多的,也是对其色谱保留性能最了解和熟悉的,在C18柱上获得的信息我们可以预测其极性,以及为解决遇到的问题问题下一步可能将采取的措施。 III)填料粒径、色谱柱型号的选择:在没有特别指明之前,最好使用我们常用的5um、4.6×150mm或250mm,和前面首选C18作初始条件一样,是为了方便预测其保留性能。3)检测器和波长的选择 目前使用最为普遍的是UV检测器,因此,在不了解其是否有紫外吸收的情况下,我们先要了解其这一性能,用标准品配成合适的溶液进行紫外扫描,收集目标化合物最大吸收波长的数据,确定波长;或者是在有DAD检测器的条件下进几针标准品溶液,通过DAD的三维谱图可以获得化合物最大吸收波长的相关信息。如无紫外吸收,则需选用合适的检测器。因为检测器是我们监测化合物是否出峰的工具,是我们的能看到化合物的“眼睛”,也是我们开发方法的基础,因此非常重要,需认真选择。[font=Times New
[color=#00008B](一)有机合成中展开剂的选择 做有机合成时走板子是常有的事,展开剂的选择就至关重要了。选择适当的展开剂是首要任务.一般常用溶剂按照[/color][color=#00FFFF][color=#DC143C]极性从小到大的顺序排列大概为:石油迷己烷苯乙醚THF乙酸乙酯丙酮乙醇甲醇[/color][/color]使用单一溶剂,往往不能达到很好的分离效果,往往使用混合溶剂通常使用一个高极性和低级性溶剂组成的混合溶剂,高极性的溶剂还有增加区分度的作用,展开剂的比例要靠尝试.一般根据文献中报道的该类化合物用什么样的展开剂,就首先尝试使用该类展开剂,然后不断尝试比例,直到找到一个分离效果好的展开剂。展开剂的选择条件:①对的所需成分有良好的溶解性;②可使成分间分开;③待测组分的Rf在0.2~0.8之间,定量测定在0.3~0.5之间;④不与待测组分或吸附剂发生化学反应;⑤沸点适中,黏度较小;⑥展开后组分斑点圆且集中;⑦混合溶剂最好用新鲜配制。 一般来说,[color=#00FFFF][color=#DC143C]弱极性溶剂体系的基本两相由正己烷和水组成[/color][/color],再[color=#DC143C]根据需要加入甲醇、乙醇,乙酸乙酯来调节[/color]溶剂系统的极性,以达到好的分离效果,适合于[color=#DC143C]生物碱、黄酮、萜类等的分离[/color]; [color=#DC143C]中等极性的溶剂体系由氯仿和水基本两相组成[/color],由[color=#DC143C]甲醇、乙醇,乙酸乙酯等来调节,适合于蒽醌、香豆素,以及一些极性较大的木脂素和萜类的分离[/color]; [color=#DC143C]强极性溶剂,由正丁醇和水组成,也靠甲醇、乙醇,乙酸乙酯等来调节,适合于极性很大的生物碱类化合物的分离[/color]。 很多时候,展开剂的选择要靠自己不断变换展开剂的组成来达到最佳效果。我们在实验中,为了实现一个配体与其他杂质有效分离,曾经尝试了很多种的溶剂组合,最后才找到石油醚—EtOAc—HCOOH(5.5:3.5:0.1)混合溶剂。一般把[color=#DC143C]两种溶剂混合时,采用高极性/低极性的体积比为1/3的混合溶剂,如果有分开的迹象,再调整比例(或者加入第三种溶剂),达到最佳效果;如果没有分开的迹象(斑点较“拖”),最好是换溶剂[/color]。对于[color=#DC143C]在硅胶中这种酸性物质上易分解的物质,在展开剂里往往加一点点三乙胺,氨水,吡啶等碱性物质来中和硅胶的酸性。[/color](选择所添加的碱性物质,还必须考虑容易从产品中除去,氨水无疑是较好的选择。) 分离效果的好坏和所用硅胶和溶剂的质量很有关系:不同厂家生产的硅胶可能含水量以及颗粒的粗细程度,酸性强弱不同,从而导致产品在某个厂家的硅胶中分离效果很好,但在另一个厂家的就不行。溶剂的含水量和杂质含量对分离效果都有明显的影响。温度,湿度对分离效果影响也很明显,在实验中我们发现有时同一展开条件,上下午的Rf截然不同 。 展开剂的选择主要根据样品的极性、溶解度和吸附剂的活性等因素来考虑,在进行薄层层析时,首先应该知道未知化学成分的类型,其极性的大致归属,从提取液或从色谱柱的流动相极性可知,另外某样品里含多种化学成分先按极性不同大致分,然后细分,对于分离未知的化学物质,展开剂的选择也是一个摸索的过程,不应该仅仅从展开剂考虑,多因素综合衡量!溶剂:层析过程中溶剂的选择,对组分分离关系极大。在柱层析时所用的溶剂(单一剂或混合溶剂)习惯上称洗脱剂,用于薄层或纸层析时常称展开剂。洗脱剂的选择,须根据被分离物质与所选用的吸附剂性质这两者结合起来加以考虑在用极性吸附剂进行层析时,[color=#00008B][color=#DC143C]当被分离物质为弱极性物质,一般选用弱极性溶剂为洗脱剂;被分离物质为强极性成分,则须选用极性溶剂为洗脱剂。[/color][/color]如果对某一极性物质用吸附性较弱的吸附剂(如以硅藻土或滑石粉代替硅胶),则洗脱剂的极性亦须相应降低。在柱层操作时,被分离样品在加样时可采用于法,亦可选一适宜的溶剂将样品溶解后加入。溶解样品的溶剂应选择极性较小的,以便被分离的成分可以被吸附。然后渐增大溶剂的极性。这种极性的增大是一个十分缓慢的过程,称为“梯度洗脱”,使吸附在层析柱上的各个成分逐个被洗脱。如果极性增大过诀(梯度太大),就不能获得满意的分离。溶剂的洗脱能力,有时可以用溶剂的介电常数(ε)来表示。介电常数高,洗脱能力就大。以上的洗脱顺序仅适用于极性吸附剂,如硅胶、氧化铝。对非极性吸附剂,如活性炭,则正好与上述顺序相反,在水或亲水住溶剂中所形成的吸附作用,较在脂溶性溶剂中为强。 被分离物质的性质 被分离的物质与吸附剂,洗脱剂共同构成吸附层析中的三个要素,彼此紧密相连。在指定的吸附剂与洗脱剂的条件下,各个成分的分离情况,直接与被分离物质的结构与性质有关。对极性吸附剂而言,成分的极性大,吸附住强。当然,中草药成分的整体分子观是重要的,例如极性基团的数目愈多,被吸附的住能就会更大些,在同系物中碳原子数目少些,被吸附也会强些。总之,只要两个成分在结构上存在差别,就有可能分离,关键在于条件的选择。要根据被分离物质的性质,吸附剂的吸附强度,与溶剂的性质这三者的相互关系来考虑。首先要考虑被分离物质的极性。如被分离物质极性很小为不含氧的萜烯,或虽含氧但非极性基团,则需选用吸附性较强的吸附剂,并用弱极性溶剂如石油醚或苯进行洗脱。但多数中药成分的极性较大,则需要选择吸附性能较弱的吸附剂(一般Ⅲ~Ⅳ级)。采用的洗脱剂极性应由小到大按某一梯度递增,或可应用薄层层析以判断被分离物在某种溶剂系统中的分离情况。此外,能否获得满意的分离,还与选择的溶剂梯度有很大关系。现以实例说明吸附层析中吸附剂、洗脱剂与样品极性之间的关系。如有多组分的混合物,象植物油脂系由烷烃、烯烃、舀醇酯类、甘油三酸醋和脂肪酸等组份。如对于C-27甾体皂甙元类成分,能因其分字中羟基数目的多少而获得分离:将混合皂甙元溶于含有5%氯仿的苯中,加于氧化铝的吸附柱上,采用以下的溶剂进行梯度洗脱。如改用吸附性较弱的硅酸镁以替代氧化铝,由于硅酸镁的吸附性较弱,洗脱剂的极牲需相应降低,亦即采用苯或含5%氯仿的苯,即可将一元羟基皂甙元从吸附剂上洗脱下来。这一例子说明,同样的中草药成分在不同的吸附剂中层析时,需用不同的溶剂才能达到相同的分离效果,从而说明吸附剂、溶剂和欲分离成分三者的相互关系。 (二)簿层层析:薄层层析是一种简便、快速、微量的层析方法。一般将柱层析用的吸附剂撒布到平面如玻璃片上,形成一薄层进行层析时一即称薄层层析。其原理与柱层析基本相似。 1.薄层层析的特点:薄层层析在应用与操作方面的特点与柱层析的比较。 2.吸附剂的选择:薄层层析用的吸附剂与其选择原则和柱层析相同。主要区别在于薄层层析要求吸附剂(支持剂)的粒度更细,一般应小于250目,并要求粒度均匀。用于薄层层析的吸附剂或预制薄层一般活度不宜过高,以Ⅱ~Ⅲ级为宜。而展开距离则随薄层的粒度粗细而定,薄层粒度越细,展开距离相应缩短,一般不超过10厘米,否则可引起色谱扩散影响分离效果。 3.展开剂的选择:薄层层析,当吸附剂活度为一定值时(如Ⅱ或Ⅲ级),对多组分的样品能否获得满意的分离,决定于展开剂的选择。中草药化学成分在脂溶性成分中,大致可按其极性不同而分为无极性、弱极性、中极性与强极性。但在实际工作中,经常需要利用溶剂的极性大小,对展开剂的极性予以调整。[/color][/color][/color][/color]
薄层色谱法的展开时间怎么选择
芳磺酸的薄层色谱应该选什么样的展开剂呢另外我用甲醇和正庚烷体系,展开后不成斑点而是散开的一片,是不是因为两者互溶性太差的问题?有经验说芳磺酸极性太大加冰醋酸能消除拖尾,但似乎不起作用,加氨水反而拖尾现象减轻,谁能解释一下为什么呢?谢了
这篇文献是天津师范大学化学系白桦等人的文章,摘要:本文通过对实验室回收的薄层色谱展开剂的再次实验所得数据与新配制的展开剂实验所得数据进行比较,指出薄层色谱展开剂是可以再次利用,且分离效果较好。这样不仅可以节省实验经费,而且有利于环境保护,减少环境污染。
选择展开剂,要依据溶剂极性和他们的混溶性,溶剂对被分析物的溶解性,以及被分析物的结构。这里只讨论药典里通常使用的以硅胶为固定相主体的正相薄层,也不考虑板的活性。 列出溶剂极性参数表,方便以下比较展开剂。环已烷:-0.2、石油醚(Ⅰ类,30~60℃)、石油醚(Ⅱ类,60~90℃)、正已烷:0.0、甲苯:2.4、二甲苯:2.5、苯:2.7、二氯甲烷:3.1、异丙醇:3.9、正丁醇:3.9、四氢呋喃:4.0、氯仿:4.1、乙醇:4.3、乙酸乙酯:4.4、甲醇:5.1、丙酮:5.1、乙腈:5.8、乙酸:6.0、水:10.2 。 关于溶剂混溶性,一般根据相似相溶原则,需要注意,极性相差大的不混溶,比如正己烷与甲醇。多元展开剂,主体的两种溶剂不能混溶,就需要通过第三种溶剂来调和。比如:石油醚、正庚烷、正已烷、戊烷、环已烷和甲醇、水之类的。 一般正相色谱,固定相为极性,被分析物质的极性越大,需要极性更大的展开剂。 了解被分析物的极性可以通过分析其结构获得,很难获得它的极性指数。物质分子化学结构中,通常由较极性部分和非极性部分两部分。例如下面以苯丙烷为极性小部分,随着极性基团部分的增加,总体的极性就增加,展开剂极性也增加了。 , 依次为肉桂酸、阿魏酸、咖啡酸、菊苣酸、绿原酸。 相应展开剂分别为:正己烷—乙醚—冰醋酸 (5:5:0.1)、苯-冰醋酸-甲醇(30:1:3)、氯仿-甲醇-甲酸(9:1: 0.5)、石油醚-乙酸乙酯-甲酸(3:6: 1)、醋酸丁酯-甲酸-水(7:2.5:2.5)。(由于薄层板、比移值不同的原因,展开剂极性比较是相对的,并非绝对的后者大于前者)。 vSf"sTT@w 现在最重要的问题是,不同化合物,怎么定它的极性,又用什么标准来定它对应的展开剂呢?以下分开讨论不同化合物极性情况及其对应的展开剂。 首先是极性较小的挥发性物质。比如:冰片:石油醚 (30~60℃)—醋酸乙酯(17:3)、厚朴酚:苯-醋酸乙酯(9:1.5)、α-香附酮:苯-醋酯乙酯-冰醋酸(92:5:5)、丹皮酚:环己烷-醋酸乙酯(3:1),这类化合物,以石油醚、正构烷和苯为体积百分数比较大的溶剂,通常起溶解和分离化合物的作用,而用醋酸乙酯为调节Rf(比移值)的溶剂。为了减少拖尾之类其他相似相溶原则以外的影响,适当加入添加剂,如有机酸或者有机碱。 极性较小的不挥发性物质。比如:β -谷甾醇:环己烷-醋酸乙酯-甲醇(6:2.5:1)或者环己烷-丙酮(5:2) 、熊果酸:甲苯-醋酸乙酯-冰醋酸(12:4:0.5)、齐墩果酸:氯仿-甲醇(40:1)、猪去氧胆酸:氯仿-乙醚-冰醋酸(2:2:1)、大黄素:苯—醋酸乙酯—甲醇(15:2:0.2)或者苯—乙醇 (8:1)、丹参酮ⅡA:苯-醋酸乙酯-甲酸(40:25:4) 、穿心莲内酯:氯仿-无水乙醇(9:1)、靛玉红、靛蓝氯仿-乙醇(9:1)或者苯-氯仿-丙酮(5:4:1)。这类物质展开剂极性比极性较小的挥发性物质洗脱力强一些,因为这类物质极性小的母核大,而极性大的基团通常可以形成氢键,比如羧酸、羟基。以上物质,母核分子量减小、母核结构中不饱和健的增加(尤其是出现苯环),极性基团的增加,都使极性增加,展开剂极性也增大。这个范围内的物质很多,一般展开剂大百分数的溶剂可以从环己烷—〉甲苯—〉二甲苯—〉苯—〉氯仿的顺序,按照极性要求选择。这里注意,异丙醇、正丁醇极性指数也比较小,在这范围的化合物很少用,因为粘性大、展开慢,造成斑点扩散;另外,羟基的氢键作用力也有不利。调节Rf值的溶剂,从醋酸乙酯—〉甲醇—〉丙酮—〉乙醇。挥发性物质也有很多带羰基、羟基的,但从它的挥发性就可以明白,分子间作用力不强,另外,母核与石油醚、正构烷和苯的结构差异小,估计更容易脱离硅胶吸附,更快进入溶剂中,而不需要通过提高展开剂的极性。 皂苷类。人参皂苷:氯仿-甲醇-水 (65:35:10)10℃以下放置的下层溶液或正丁醇-醋酸乙酯-水(4:1:5)的上层溶液或氯仿-醋酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液、芍药苷:氯仿-醋酸乙酯-甲醇-甲酸(40:5:10:0.2)、黄芩苷:醋酸乙酯-丁酮-醋酸-水(10:7:5:3)、橙皮苷:苯—醋酸乙酯—甲酸—水(1:12:2.5:3)的上层溶液、葛根素:氯仿-甲醇-水(14:5:0.5)、芦丁:醋酸乙酯-甲酸-水(8:1:1)。这类物质,由于存在糖的多羟基结构,苷元的结构影响变小。展开剂中使用极性大的有机溶剂(氯仿、醋酸乙酯、甲醇、正丁醇)和水。乙酸和甲酸的使用,一方面增大展开剂极性,另外也可以抑制硅胶羟基的作用,减少拖尾。由于混溶性和硅胶耐酸能力的限制,水和酸的使用是有限度的。 极性大的小分子有机酸。没食子酸:氯仿-醋酸乙酯-甲酸 (5:4:1)、阿魏酸、咖啡酸、菊苣酸、绿原酸、异绿原酸。这类物质多数是苯乙烯母核的,这个结构的极性本身比较大,另外有酚羟基和羧酸基团,个别有多羟基配基。皂苷的展开剂差不多,极性大。注意甲酸通常指的是浓度85%左右的,含有水。 含氮有机物。盐酸小檗碱:苯-醋酸乙酯-甲醇-异丙醇-浓氨试液(12:6:3:3:0.6)(氨蒸气饱和) 或正丁醇-冰醋酸-水(7:1:2)、麻黄碱:氯仿-甲醇-浓氨试液(20:5:0.5)或正丁醇-冰醋酸-水(8:2:1)、甘草酸铵:醋酸乙酯-甲酸-冰醋酸-水(15:1:1:2)。由于NH2硅醇基的作用很强,在强极性展开剂加有机酸、有机碱扫尾。对于极性化合物,使用正丁醇对斑点扩散影响较小,因为化合物和硅胶的作用强。 进行薄层分析基本可以根据母核、基团,选择相似的化合物对号入座。当然,具体的条件优化则需要根据实际情况了。遇到较困难的分离,需要使用到设计优化方法的,已经不属于本文讨论范围了。
求有关光谱类的外文文献网址或者镜像
求有关光谱类的外文文献网址或者镜像







 我要推广仪器
我要推广仪器
 下载APP
下载APP