色谱级正丁醇杂质峰与乙醇峰分不开打正丁醇纯物质出现3个峰 主峰为正丁醇 另外两个排除进样问题,可以确认是杂质峰。本人使用的是SP6890色谱仪,FFAP色谱柱,FID检测器,程序升温,内标法定量。以正丁醇为溶剂,乙醇为溶质,萘为内标物配置标准溶液。发现乙醇峰与正丁醇的一个杂质峰重叠较为严重无法忽略,影响最后的定量。试过将柱室温度从80降到40度 保留时间从3min延长至10min,柱头压从0.05降低至0.037,还是很难分离。
如果GC不能将2种物质分开,2种物质的峰是重叠的,但这2种物质定量离子不同,质谱会根据定量离子来积分的,这样做应该可以吧?定性的话可以通过峰纯度来看
如题,你们在进行定性定量分析时候,发现基线漂移,是如何进行校正的?另外,定性定量分析时候,也经常遇到重叠色谱峰,这个时候你们是怎样处理的?谢谢指教
我是一名[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]新手,现在根据国标在重新做苯系物 的标线,但是色谱峰出峰时间都提前了很多,原来8.5分钟出完,现在5分钟就出完,而且少了一个,是有两个峰重叠了,降低了柱温,没有效果,请各位老师指点一下,用的是KB-WAX的柱子
完全重叠的2个峰 除了sim定量 如果用普遍的校正因子发定量 面积一定不准 大家用什么方法又比如溶剂大风有其他化合物共流出 怎么定量

进样重叠造成的色谱峰分叉 概述:由于多次进样造成的色谱峰分叉现象 日前在实验室做气体分析的时候,不慎发生了进样重叠的现象,即进行了两次间隔时间很短的进样操作,得到的谱图颇有点意思,稍做粗略分析。 在实验室使用了Shimadzu的GC-2010plus + MGS-4手工气体进样阀,进样某混合烃类气体,样品气保存在气袋中,柱温采用了程序升温。 MGS-4阀有两个位置,如图左为取样位置(charge或者load),在此状态下,将样品气体通入in端,样品被保留在定量环中。图右为进样位置(discharge或者inject),此状态下,样品气体进入GC系统。http://ng1.17img.cn/bbsfiles/images/2014/11/201411262207_524758_1604036_3.jpg 第一次进样,得到下图中红色的谱图。在做第二次进样的时候,不小心提前误转动了进样阀,然后迅速恢复load位置,重新进样。此时由于气袋是连接到进样阀上的,应该有微量扩散的样品存在进样阀中,错误转动阀,相当于进样。那么第二次进样分析,实际是执行了两次进样操作。 第二次进样的谱图即下图中的黑色部分。 可以看出,11min的色谱峰出现了分叉,其实是两次进样造成的。随着分析时间的延长,分叉现象逐渐减弱,在16min附近的色谱峰分叉要轻微的多,在17min的色谱峰,分叉现象基本消失。 http://ng1.17img.cn/bbsfiles/images/2014/11/201411262208_524759_1604036_3.jpg http://ng1.17img.cn/bbsfiles/images/2014/11/201411262208_524760_1604036_3.jpg 该现象的原因颇值得讨论,这个现象非常类似液体样品溶剂聚焦条件不良得到的谱图。 对于气体样品,原因较为类似,样品进入色谱柱中后,在一定的柱长度上分布和保留,这个分布和保留是在不断被载气推动的情况下发生的,那么由于固定相聚焦的原因,样品分布会逐渐变窄(结果类似溶剂聚焦)。 两次进样,样品分布在色谱柱内的长度会变长,色谱柱内同时存在样品分布空间差和聚焦两个现象。保留时间短的组分,聚焦不足以补偿样品分布问题,就会产生双峰。保留时间长的组分则反之,所以会得到没有分叉的色谱峰。 参见 http://bbs.instrument.com.cn/shtml/20121126/4391256/ 小结:还是聚焦问题。[/

色谱峰重叠,改如何算峰面积?如图所示[img=,690,1226]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071322_01_3227746_3.jpg[/img][img=,690,517]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071322_02_3227746_3.jpg[/img]
液相色谱中峰重叠 如何使其分开
各位老师学长,想向大家请教一个问题。我在看《高等结构分析》时,看到这么一段话“红外光谱定量方法主要测量谱带的强度和测量谱带的面积两种。此外也有谱带的一阶和二阶导数的计算方法,这种方法能准确地测量重叠的谱带,甚至包括强峰斜坡上的肩峰。”想请问这种求导数方法是怎么实现定量的?其原理是什么?具体的操作步骤是那些?非常感谢![em01]
色谱级正丁醇杂质峰与乙醇峰分不开。打正丁醇纯物质出现3个峰 主峰为正丁醇 另外两个排除进样问题,可以确认是杂质峰。本人使用的是SP6890色谱仪,FFAP色谱柱,FID检测器,程序升温,内标法定量。以正丁醇为溶剂,乙醇为溶质,萘为内标物配置标准溶液。发现乙醇峰与正丁醇的一个杂质峰重叠较为严重无法忽略,影响最后的定量。试过将柱室温度从80降到40度 保留时间从3min延长至10min,柱头压从0.05降低至0.037,还是很难分离。求大家指教!感激不尽!
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。 可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可点击头像联系
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可联系我 电话:13926563756 qq:648048428 Email: midstone@126.com
[size=3] 本人用的是安捷伦7890A配置的色谱工作站,样品连续进样6针,跑出来的色谱峰几乎能重叠在一起,但是由于各色谱图中基线高低的微小差异,而使6针之间峰面积的RSD有时候会大于5%。我主要是调节了积分参数中的斜率灵敏度,结果没有什么改善。请问各位高手,如何设置色谱工作站的积分参数,使积分结果更准确。或者是如何设置色谱工作参数,使色谱图的基线保持一致?用基线补偿功能能行吗,如何使用?谢谢![/size]
两种标准品,色谱峰有重叠,请问有什么方法可以分离吗?我不想改变流动相的配比,改变流速是不是可以呢?[em0906]柱温35,254nm,流动相甲醇:乙酸=45:0.2%
各位大神,最近在做苯系物,采用的是岛津气质2010ultra,邻二甲苯和间二甲苯出峰重叠,并且两个物质的定量离子都是91,我该如何分别对两者进行准确定量呢,我之前用安捷伦的仪器,可以通过定量离子和定性离子的不同比例来提却离子,并分别定量,岛津的仪器怎么处理呢?还有岛津的仪器在分析样品时不可以SCAN和SIM一起做吗?
[color=#444444]测精油里边的挥发性物质,在测定物质时发现丙酮峰与其他物质峰重叠了,怎么分离开呢?[/color][color=#444444]实验用的是顶空进样,具体操作如下:称量一定量的物质于样品瓶中,一定温度下顶空进样。[/color][color=#444444]仪器条件设置如下:[/color][color=#444444]使用的是HP-FFAP毛细管柱,30.0m*0.25mm*0.25μm;溶剂使用的是四氢呋喃;[/color][color=#444444]程序升温:40℃(3 min),以3.5℃/min的速率升到65℃(0 min),最后以20℃/min的速率升到220℃(5 min)。进样口温度为240℃,丙酮在2.324min出峰,因为用的是GC,只有依靠保留时间来进行定性,有个不知名物质峰和它重叠在一起。怎么把和它离得近的不知名物质峰分离开来,或者把丙酮峰往后移一下。使用程序升温或者换溶剂、柱子管用否?[/color][color=#444444]路过的各位大神,给解决一下哈![/color]
乙炔和乙烯用色谱峰老是重叠怎么办呀
[b][color=#444444]谱图中重叠峰是垂直分割积分好,还是用其他方法来进行积分好?[/color][/b]
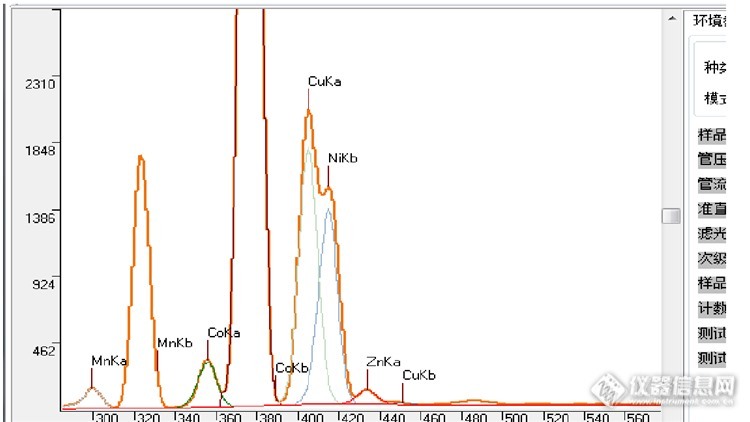
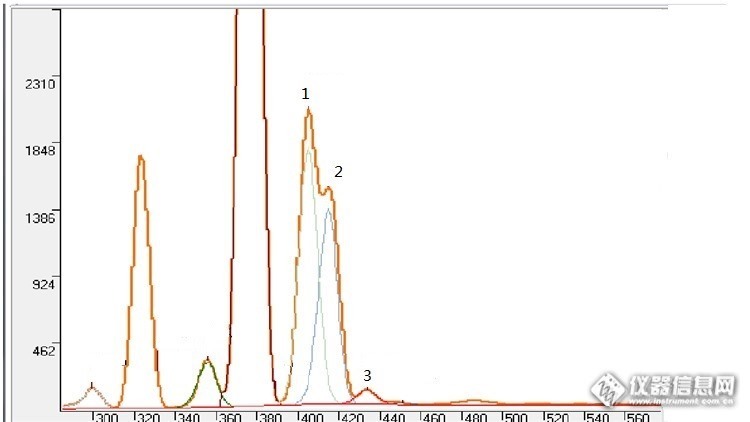
元素的峰重叠在光谱分析中对定量结果影响很大,需要进行分离。重叠峰分离的方法有很多,比如:傅立叶变换、偏最小二乘法等。这里介绍一种卡尔曼滤波算法。卡尔曼滤波器是一个“optimal recursive data processing algorithm(最优化自回归数据处理算法)”。对于解决很大部分的问题,他是最优,效率最高甚至是最有用的。他的广泛应用包括机器人导航,控制,传感器数据融合甚至在军事方面的雷达系统以及导弹追踪等等。近年来更被应用于计算机图像处理,例如头脸识别,图像分割,图像边缘检测等等。具体的算法,因为极其复杂,所以不在这里罗列,感兴趣的可以去搜索相关资料。这里演示一下在用能散XRF测镍铜合金中的一个案例:http://ng1.17img.cn/bbsfiles/images/2017/03/201703051157_01_1780790_3.png图中镍的Kb峰、铜的Ka峰和锌的Ka峰、铜的Kb峰存在重叠。谱峰分离效果如图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力只能回归到分支比扣除法。
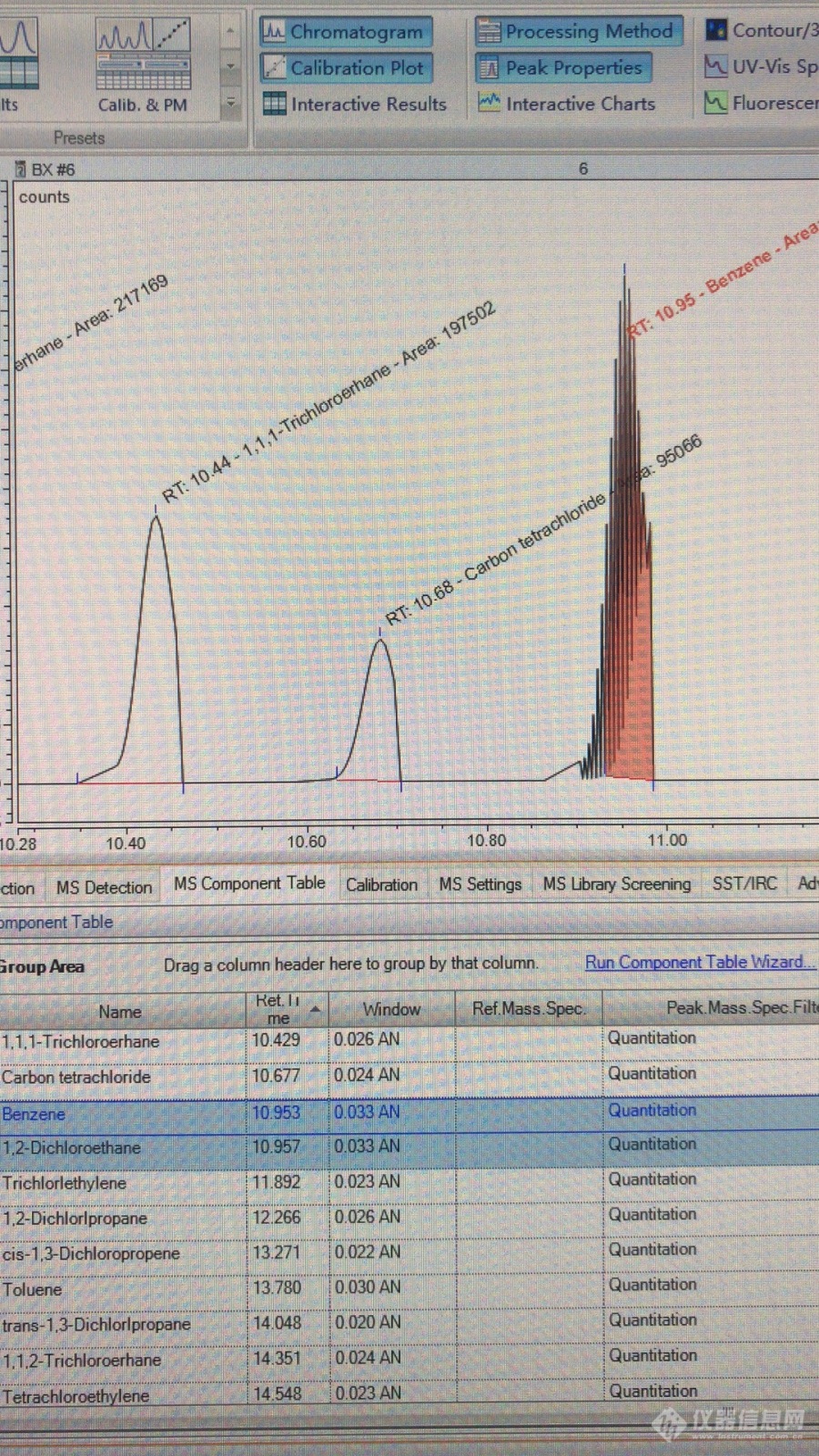
公司最近新买了一台GCMS,小弟按照国标HJ644-2013做标准系列的时候发现有几个峰是重叠的,下图中是1,1-二氯乙烷和苯峰重叠在一起,培训老师跟我们说重叠在一起也没关系只需要用特征离子扫描还是可以定性定量,但是图谱上出现锯齿状的形态,用时间光谱工具点击峰发现峰内的是一种物质,峰外的是另一种物质,软件是变色龙!各位老师帮小弟看看软件上哪里可以调整或者其他的解决方法。[img=,690,1226]https://ng1.17img.cn/bbsfiles/images/2019/05/201905292026139354_4925_2648031_3.jpg!w690x1226.jpg[/img]
用分子筛做苯酚羟基化反应时,用安捷伦1100高效LC测试,发现反应中所用的溶剂丙酮与产物对苯二酚峰有很大重叠,虚心请教大家该怎么处理?是柱子的原因呢,还是参数(流动相,柱温等)没设好?用的是ZORBAX Eclipe C18-XDB柱子,我刚接触液相色谱,希望高手指点。

最近在做邻苯二甲酸脂,标样是16脂的,出峰就只有15个峰,DHXP与DBP的峰重叠在一起了,各位有这样的情况吗?怎么定量啊?http://ng1.17img.cn/bbsfiles/images/2012/02/201202241715_350938_2345710_3.jpg
我们常用色谱图重叠来进行图谱对比。常用的重叠图,我们都大概用这样的,如图一。看起来也是清晰,但需要拉动看没有峰型差异。我们也可以试试另外一个重叠色谱图方法,如图二。通过上下错开来进行对比。可以直接看到丰度进行比较。大伙们,你更喜欢哪种看图模式呢??[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111251211138607_2408_3253514_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111251211140960_2545_3253514_3.png[/img]
采用内参比定转化率时,出现特征峰与参比峰的重叠,是否能用高斯函数拟合的方式将重叠峰分离,然后根据修正后的结果进行计算?或当遇到此类情况的时候有无其他解决办法?
面积归一法、内标法、外标法、标准加入法。这么多定量法,用哪一种呢?每一种的适用范围是什么呢?其优缺点又是什么?其标准物又有什么要求呢?看完你就都知道了。归一化法把所有出峰的组分含量之和按100%计的定量方法,称为归一化法。各成分校正因子一致时可用该法,该法简便、准确,特别是进样量不容易准确控制时,进样浓度及进样量的变化的影响很小。其他操作条件,如流速、柱温等变化对定量结果的影响也很小。GC应用广于HPLC。外标法(标准曲线法、直接比较法)首先用欲测组分的标准样品绘制标准工作曲线。具体作法是:用标准样品配制成不同浓度的标准系列,在与欲测组分相同的色谱条件下,等体积准确量进样,测量各峰的峰面积或峰高,用峰面积或峰高对样品浓度绘制标准工作曲线,此标准工作曲线应是通过原点的直线。若标准工作曲线不通过原点,说明测定方法存在系统误差。标准工作曲线的斜率即为绝对校正因子。当欲测组分含量变化不大,并已知这一组分的大概含量时,也可以不必绘制标准工作曲线,而用单点校正法,即直接比较法定量。单点校正法实际上是利用原点作为标准工作曲线上的另一个点。因此,当方法存在系统误差时(即标准工作曲线不通过原点),单点校正法的误差较大。因此规定,y=ax+b 。b的绝对值应不大于100%响应值是y的2%。标准曲线法的优点:绘制好标准工作曲线后测定工作就很简单了,计算时可直接从标准工作曲线上读出含量,这对大量样品分析十分合适。特别是标准工作曲线绘制后可以使用一段时间,在此段时间内可经常用一个标准样品对标准工作曲线进行单点校正,以确定该标准工作曲线是否还可使用。标准曲线法的缺点:每次样品分析的色谱条件(检测器的响应性能,柱温度,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。另外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),因此对样品前处理过程中欲测组分的变化无法进行补偿。内标法选择适宜的物质作为欲测组分的参比物,定量加到样品中去,依据欲测组分和参比物在检测器上的响应值(峰面积或峰高)之比和参比物加入的量进行定量分析的方法称为内标法。内标法的关键是选择合适的内标物。内标物应是原样品中不存在的纯物质,该物质的性质应尽可能与欲测组分相近,不与被测样品起化学反应,同时要能完全溶于被测样品中。内标物的峰应尽可能接近欲测组分的峰,或位于几个欲测组分的峰中间,但必须与样品中的所有峰不重叠,即完全分开。一般会选择标准物质的同位素物质作为内标物。内标法的优点:进样量的变化,色谱条件的微小变化对内标法定量结果的影响不大,特别是在样品前处理(如浓缩、萃取,衍生化等)前加入内标物,然后再进行前处理时,可部分补偿欲测组分在样品前处理时的损失。若要获得很高精度的结果时,可以加入数种内标物,以提高定量分析的精度。内标法的缺点:选择合适的内标物比较困难,内标物的称量要准确,操作较麻烦。使用内标法定量时要测量欲测组分和内标物的两个峰的峰面积(或峰高),根据误差叠加原理,内标法定量的误差中,由于峰面积测量引起的误差是标准曲线法定量的2-2,但是由于进样量的变化和色谱条件变化引起的误差,内标法比标准曲线法要小很多,所以总的来说,内标法定量比标准曲线法定量的准确度和精密度都要好。标准加入法标准加入法实质上是一种特殊的内标法,是在选择不到合适的内标物时,以欲测组分的纯物质为内标物,加入到待测样品中,然后在相同的色谱条件下,测定加入欲测组分纯物质前后欲测组分的峰面积(或峰高),从而计算欲测组分在样品中的含量的方法。标准加入法的优点:不需要另外的标准物质作内标物,只需欲测组分的纯物质,进样量不必十分准确,操作简单。若在样品的前处理之前就加入已知准确量的欲测组分,则可以完全补偿欲测组分在前处理过程中的损失,是色谱分析中较常用的定量分析方法。标准加入法的缺点:要求加入欲测组分前后两次色谱测定的色谱条件完全相同,以保证两次测定时的校正因子完全相等,否则将引起分析测定的误差。
如题。 双峰重叠常见的是垂直切割和拖尾处理。 要分具体情况。 多峰重叠情况如何? 欢迎讨论
我在用XRF测量合金的组成时发生谱峰重叠,导致无法得知正确的质量百分比。请问有什么解决方法吗?[color=#00008B]谢谢,换了谱线,已经能测了。你说的数学软件,如果可以请推荐一个。我用的是理研ZSX-100e[/color]
请问哪位老师做过磺胺总量?甲恶唑和二甲氧峰位置总是重叠,定量碎片离子也一样?怎么办
根据GB/T 20756-2006标准开展水产品中氯霉素测定,检测方法参数由仪器公司提供并优化,二维色谱图中显示氯霉素和氯霉素-D5出峰重叠,但可以通过离子对定性,并使用内标法定量。内标法定量核心要求是目标物和内标必须分开,请教各路大神是否需要继续优化检测方法,直至目标物和内标物分别出峰。
果葡糖浆里色谱分析出来的某个峰和糖精钠是同一时间,而且加标后就重叠了,包括蜂蜜也有个和糖精钠重叠的峰,如果蜂蜜和果葡糖浆里没有加过糖精钠,应该怎么区分呢?或者有知道用C18短柱做甜味剂,分析过果葡糖浆的大哥大姐有吗?知道和糖精钠同一时间出峰的那个峰是什么吗







 我要推广仪器
我要推广仪器
 下载APP
下载APP