我是菜鸟,现公司有一台SP6800的色谱仪,用来则烟纸中的挥发物的,当中最主要的是要控制苯的含量。原来是用面积外标法计算含量,测试中所用的测试方法是原来做的一条曲线(四级标液校正线)。2月份修了一次,现在时不时有客户投诉苯超标。现在我想问的是,(1)在原有的校正曲线上,要如何校正这条曲线?(2)我要重新做一条曲线的话,又要怎么做?怎么选择校正液,用哪一档的合适?我们苯的指标是0.003mg/m2~0.007mg/m2 ,有些客户要求不能超 0.003mg/m2 。原有的四级标液中苯的含量是0.1024mg/m2。这会不会相差太远了呢?我的想法是:对于问题(1),是不是在工作站中选择本身带的“面积外标法”先测试标准液,测完后就按”校正“就可以了呢?问题(2)也是同上,进多几个样,然后就校正就可以了呢?
[color=#444444]是这样的。我换了一根[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱,做校准曲线发现相关系数不太好,才0.965。去年用的旧色谱柱线性相关系数是0.995。但是两次做校正曲线进样体积不一样。新柱子进样体积是10,20,30,40,50ml,旧柱子进样体积是10,20,50,100ml。然后我测了一个相同的样品,发现结果基本上相同。我想这根新柱子这样的校正曲线可以用吗?是不是线性相关系数一定要达到三个9以上才行?[/color]
安捷伦[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]SCD检测器,自动进样。主要检测焦炉煤气中的硫化物。最近一直检测不出来,进了标准气,结果也偏低了好多,想重新校正曲线,是单点校正的。没接触过不知怎样校正,请老师们帮忙指点一下。
新买的7890,今天作了内标法的校正曲线,明明在“报告设定”中选择了“峰面积”,打印出来的校正曲线纵坐标是“峰面积比”,但是Y值却写着“峰高”。到底是我哪里设错了,还是软件的bug呢?
各位前辈: 我刚接触[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]。请教: 用火焰法检测食品中的锰含量时,校正曲线法是选一次线过零还是不过零,两者有什么区别?
买仪器时,仪器工程师培训我们时,在做校正曲线时都是选择”强制过原点“,仪器用了1年多,现在每次做校正曲线再选择”强制过原点“就不成线性,这会就用的”不过原点“。想问哪些因素会导致这个问题?过不过原点有何影响?
[table][tr][td]用GC做溶剂残留,需作校正曲线。如果在做校正曲线时,发现其中一点与其他四点偏离了,应该如何处理呢?是去掉该点作出校正曲线呢,还是采用其他办法,如重做校正曲线?[/td][/tr][/table]
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]使用内标法时,我们的校正曲线是浓度比个峰面积比,但是校正后的样品是真实浓度而不是浓度的比值?如何得到这个比值呢?10345中做脂类和醇时都需要这个值
1、从减小校正曲线的置信区间考虑,用4-6个实验点建立校正曲线比较合适,实验点要均匀分布;2、在校正曲线的线性范围内,要尽量扩大浓度的取值范围,且被测组分含量要位于校正曲线中间(根据统计学规律,这样误差较小);3、在总实验工作量一定时,适当增加实验点数目,减少实验点重复测量次数是有利的;增加每个实验点的重复测定次数只能提高个别实验点的测定精密度,而增加实验点数目能增加校正曲线的稳定性(从测定时单个点的RSD可以看出);4、在高浓度、低浓度两端的浓度测定响应信号的精密度比校准曲线中央区域差,适当增加它们重复测量次数以提高它们的测定精密度(多次测定,取平均值,误差较小);5、将空白溶液实验点参与回归,增加实验点数目,以提高校正曲线的稳定性;按照校正曲线的标准偏差分布曲线,在空白溶液时测定信号的标准偏差较大,因此不宜用空白溶液调零,应采用校正曲线截距扣空白,以减少零浓度点测定波动性对校准曲线定位的影响,提高空白扣除的准确度(空白测定时,RSD很大,几十到几百);6、校正曲线不得任意外延(较高浓度,校准曲线会完全,误差较大,有时候会成倍数关系的错);7、当需要对校准曲线变动性进行校正时,宜用不同于制作工作的浓度值重新标定校准曲线,以增加回归线的实验点数目,提高校正曲线的稳定性;8、不要用重新测定的个别实验点,与原校正曲线的原点连接直线(即重置斜率),或通过个别点将曲线平移的方法使校准曲线变动,最好将原实验点与新测定的实验点并和,重新建立校准曲线(测定两次校准曲线点,你会发现浓度是有区别的)。
比如说做个校正曲线,5个点分别是1mg/ml,2mg/ml,3mg/ml,4mg/ml,5mg/ml,那么我分别称取这5个浓度点做的校正曲线,和我称取一个点,然后依次稀释做出的校正曲线,两者的区别在哪里?求大师指点啊?
发现工作站做的校正曲线和用excel做的校正曲线计算出来结果稍有差别?请问是什么原因呢?您遇到过这种情况吗?
盐酸利多卡因注射剂药典标准:有关物质:精密量取本品适量,用流动相定量稀释制成每1ml中约含盐酸利多卡因2㎎的溶液,作为供试品溶液;精密量取1ml,置100ml量瓶中,用流动相稀释至刻度,作为对照溶液;另取2,6二甲基苯胺对照品,精密称定,加流动相溶解并稀释制成每1ml中约含0.8微克的溶液,作为对照品溶液。照含量测定项下的色谱条件,取对照溶液20微升,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%;再精密量取上述三种溶液各20微升,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3.5倍,供试品溶液的色谱图中如有与2,6-二甲基苯胺保留时间一致的色谱峰,其峰面积不得大于对照品溶液主峰面积(0.04%),其它单个杂质峰面积不得大于对照溶液主峰面积的0.5倍(0.5%),其它各杂质峰面积的和不得大于对照溶液主峰面积(1.0%)。问题:1、请问上面的使主成分色谱峰的峰高约为满量程的20如何理解?我今天测的对照品溶液的主峰为0.0019,按百分之20,我把Y轴调到0.0095,但是在测供液时其主峰高为0.17,量程不够,请问怎么办?谢谢2、还有我在测供试液时由于上面说的我把量程调到了0.0095,主峰高大于量程色谱图在主峰超出量程前出现中断(在第2分钟时有一小段没有曲线),并且此图不能积分,请问这是为什么怎么解决?谢谢求高手指点!!!3、岛津LC-2010C怎样建立校正曲线?(具体操作)(在说明书里没有说明)谢谢!!!
我们现在是用丙酮和正丁醇做校正曲线的,一共是7个浓度值,每个浓度做三次,我想请教一下,在做校正曲线时以一个浓度的第一个值设定级别后,如何将另外两个相同浓度的峰值加进去以计算平均值?我的同事教我用校正“添加峰”添加另外两个值,但是我发现好像不对,我按他说的做了以后相对应的值还是第一个浓度的值,请求帮助,谢谢!

[b][font='Times New Roman'][font=宋体]问题描述:利血平分子量[/font]608[font=宋体],为什么在排阻色谱实验过程中,实验校正曲线上相对应的[/font][/font][font=宋体]是[/font][font='Times New Roman'][font=宋体]分子量[/font]400[font=宋体]多的洗脱体积?[/font][/font][font='Times New Roman'][font=宋体]解答:[/font][/font][/b][font=宋体][font=宋体]([/font]1[font=宋体])[/font][/font][font='Times New Roman'][font=宋体]排阻色谱不仅跟目标物的分子量大小有关,跟目标物的空间结构也有很大的关系,空间结构越紧密,相比之下洗脱顺序就越靠后[/font][/font][font=宋体]。[/font][font=宋体][font=宋体]([/font]2[font=宋体])虽然[/font][/font][font='Times New Roman'][font=宋体]利血平的分子量有[/font]608[font=宋体],[/font][/font][font=宋体]但是由于[/font][font='Times New Roman'][font=宋体]空间结构紧密,[/font][/font][font=宋体]因此[/font][font='Times New Roman'][font=宋体]在校正曲线上其对应的是分子量[/font]400[font=宋体]多的洗脱体积。[/font][/font][font='Times New Roman'][font=宋体][img=,256,256]https://ng1.17img.cn/bbsfiles/images/2021/03/202103172135399756_5075_3389662_3.png!w256x256.jpg[/img][/font][/font]
请问福立GC9790Ⅱ[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的 FL9510反控工作站如何打印校正曲线?点鼠标的左右键没有反应。
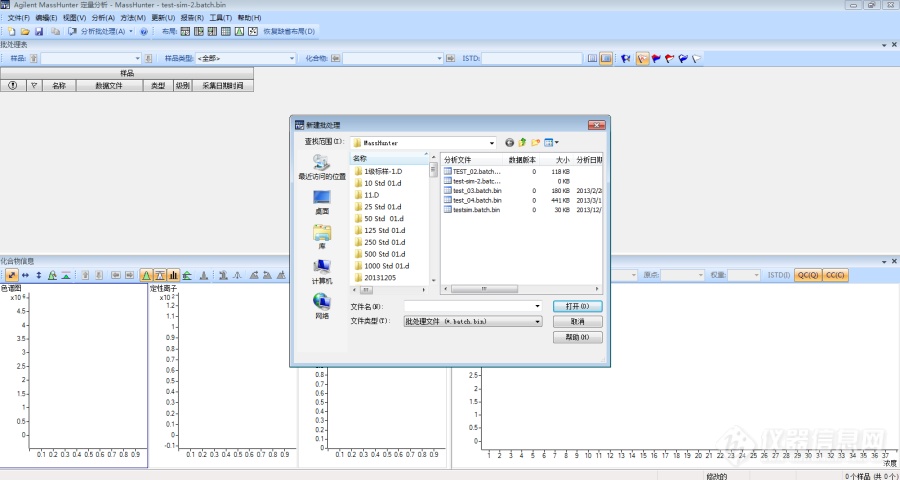
[align=center][size=18px]新手看过来,教你如何使用[/size][size=18px]Masshuner SIM[/size][size=18px]数据建立校正曲线[/size][/align]在日常分析工作中,数据处理是一个很关键的步骤,在数据处理之前,我们往往首先需要用校正标样走一下校正曲线各个浓度点,然后利用软件采集校正曲线各个浓度点的数据以此来建立我们常说的“校正曲线”。当我们遇到一个新的工作站时,往往不知道从何下手,此次以Masshuner工作站为例,给大家演示一下如何建立校正曲线。1.首先我们新建批处理,将来这个批处理中不仅存有原始数据,还包括我们后续建立的校正曲线,所有对样品的操作记录都存储在这个对应的批处理中,相当于给这一整套流程操作进行了“打包处理”,也方便了以后我们调用查看数据。[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325575436_6301_3141805_3.png[/img]2.点击菜单栏文件-添加样品,确认类型选择校正,级别处输入级别号,根据浓度从小到大依次输入1~5级别,然后选择方法菜单下编辑进入方法编辑界面。[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325578785_2326_3141805_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325579556_225_3141805_3.png[/img]3.在色谱图上鼠标右键选择色谱峰,选择后双击出现质谱图,在定量离子上鼠标点击出现蓝色箭头,鼠标右键选择新建化合物,然后输入化合物名称,在定性离子上鼠标点击出现蓝色箭头,鼠标右键选择新建定性离子。[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325582662_4223_3141805_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325584019_3535_3141805_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325585250_4590_3141805_3.png[/img]4.如果是内标法,请参考下面截图,设置好内标,需要设置的有3列(ISTD化合物名称、ISTD标记、ISTD浓度),如果是外标法,就没有这些设置步骤。[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325586188_4940_3141805_3.png[/img]5.在方法菜单下选择从校正样品创建级别,并且输入化合物浓度[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325587487_1245_3141805_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325588316_2151_3141805_3.png[/img]6.全部输入好后进行校正曲线验证,验证查看有无错误提示,如果没有错误提示那说明此次建立校正曲线成功,然后保存曲线退出并将此曲线应用于该批处理。如果验证有错误,那说明建立曲线有问题,可能是哪个地方漏输入什么参数或者漏了步骤,那就回过头来查看一遍,重复此前的操作,直至曲线验证通过。[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091325589781_5681_3141805_3.png[/img]
0 ,对NPD检测器而言更是如此。该法的几个主要要点是:a.用线性拟合得到校正曲线方程:y=ax+b……………………………… (a)注意这里的方程至少要有5 个点,每个点至少要作5次测量。根据多次测量结果可以算出,95%置信度的响应值:yx=0.095=b+t0.05•Sb……....................................(b)式中yx=0.095为95%置信度时的截距值,b为较正曲线方程中的截距,t0.05为t-分布在自由度(r)等于n-2时的单尾(one sided)t值(可查表获得,tn:5=2.353),为校正曲线截距的标准偏差(即n次测量的截距的标准差)。b. 据此算出新对应的目标分析物的最低检出限(以浓度单位表示):MDL=(yx=0.095-b)/a=t0.05Sb/a …………………………….(c)式中a 为校正曲线的平均斜率,t0.05定义与(b)式同。对禁用物质,根据欧盟规定,其a值选用0.01,以保证假阳性的检出几率在1%以内。此时的方法检出限(此处为Decision Limit,CCα)采用自由度≈∞[/fon

一直在用别人的机子(TAS-990),对软件的使用不系统。最近处理数据时,发现校正曲线详细信息界面有一个“启用误差校正功能”,勾选该选项前后,相同样品(吸光度一致)的浓度发生了不同的变化,请问大家是怎么回事儿?详见附图。http://ng1.17img.cn/bbsfiles/images/2017/03/201703292150_03_2913979_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703292150_04_2913979_3.png
光吸收的最简式A=KC,只适用于理想状态均匀稀薄的蒸汽原子,随着吸收层中原子浓度的增加,上述简化关系就不应用了。在高浓度下,分子不成比例地分解 相对于稳定的原子温度,较高浓度下给出的自由原子比率较低。(1)由于有不被吸收的辐射、杂散光的存在,不可能全部光被吸收到同一程度来保持曲线线性 (2)由于光源的老化或使用高的灯电流引起的空心灯谱线扩宽和产生自吸 (3)由于单色器狭缝太宽,则传送到检测器去的谱线不可能只有一条,校正曲线表现出更大的弯曲(4)样品中元素的电离电位不同,在火焰中发生电离时,不同元素的基态原子数不同。浓度低时,电离度大,吸光度下降多 浓度增高,电离度逐渐减小,吸光度下降程度也逐渐减小,所以引起标准工作曲线向浓度轴弯曲(下部弯曲)。
10-HDA内标法校正曲线设置怎么做啊,并且校正因子是系统自动算出来的,还是要自己另外在算啊[img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901091006158299_3338_3483704_3.jpg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901091006402362_6947_3483704_3.jpg[/img]
问题如下: 我作了5个单样的标准品谱图,但是作校正曲线时,使用一阶拟合校正曲线,有两个点偏差较大,不在校正直线上,具体可以看附件图谱。我想用二阶或者3阶等方法拟合,不过不知道怎么判断那种拟合才是最合适的,哪位高手能够指点一下??不胜感激。[~79044~]

10-HDA内标法校正曲线设置怎么做啊,并且校正因子是系统自动算出来的,还是要自己另外在算啊[img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901091006158299_3338_3483704_3.jpg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901091006402362_6947_3483704_3.jpg[/img]
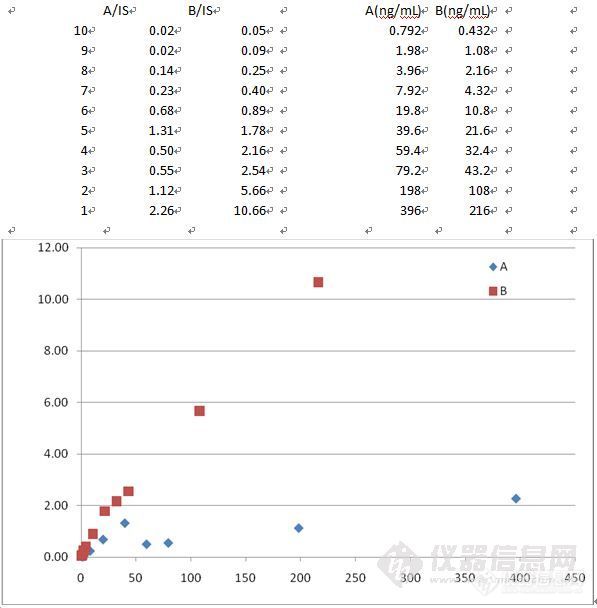
用MRM (API4000)测定A、B两个化合物在血清中的校正曲线。stock solution是A,B的混标。结果见附件。B化合物成线性,在高浓度有些饱和。A化合物在分两端成线性,但是高浓度区的线性很诡异,响应掉了下来。请高手解释。(不是stock solution的问题,因为B作出曲线了)问题终于解决了。原因是血样处理方法不对,主要是B化合物有较大的蛋白吸附(80%),在高浓度时,吸附更加严重。而我采用的是蛋白沉淀法,造成高低浓度萃取回收率有较大差异,而内标萃取回收率稳定,从而造成了这种现象。现在,在血清中加入定量的醋酸可以解吸化合物B,提高B的萃取回收率。校正曲线就正常了!http://ng1.17img.cn/bbsfiles/images/2011/03/201103051448_280952_1644952_3.jpg
校正曲线在分析中有什么作用?在什么情况需要建立?这对数据分析很重要吗?
测农残可以只做三个点的校正曲线吗?
1.大家在做液相检查时,是否每次都做校正曲线,或者多久重新建立校正曲线呢 2.我们这就对方法学做过确认,重来没有做过校正曲线啊,是否可以3.做校正曲线有什么意义呢,通过工作站计算自动得出 样品浓度,我不在工作站中建立校正曲线,在检验记录上 A1/A2=C1/C2,不就直接得到样品浓度了么 ,也不需要那么麻烦去做校正曲线
做了 一条校正曲线,校正曲线的线性很好,r=0.998,把标线上的点带入曲线计算也没问题。但是在仪器上打印定量报告时偏差就很大,请问是什么原因?
光吸收的最简式A=KC,只适用于均匀稀薄的蒸汽原子,随着吸收层中原子浓度的增加,上述简化关系不成立在高浓度下,分子不成比例地分解。结果,相对于稳定的原子温度,较高浓度下给出的自由原子比率较低(1)由于有不被吸收的辐射、杂散光。因为必须全部光被吸收到同一程度才能保持线性(2)由于光的老化或使用高的灯点流引起的空心灯谱线扩宽(3)由于单色器狭缝太宽,则传送到检测器去的谱线会超过一条。校正曲线表现出更大的弯曲
PE DRC 仪器,校正曲线类型如何选择,什么情况下选simple linear,什么情况下选thru zero?
刚上手waters液相,请问校正曲线怎麽建,谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP