由【求助】测有关物质色谱条件的专属性试验问题 http://bbs.instrument.com.cn/shtml/20101025/2882558/想到的1、两杂质色谱峰达到基线分离时的峰面积之和与两色谱峰部分重合时的峰面积之和,在使用自身对照法时它们所占的比例是一样的吗?2、用于校正归一法时,我觉得两峰应该完全分离,你觉得的呢?
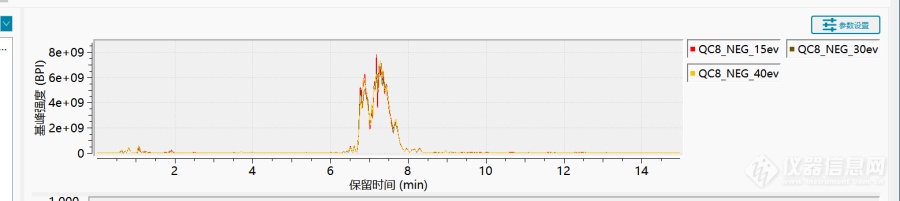
用海参做代谢,仪器是thermo的Q E ,得到的色谱图好像都没有分离,全聚在中间成一坨,是什么原因呢,样品处理时用的是鲜海参,会不会是因为盐含量高引起的?[img=,690,154]https://ng1.17img.cn/bbsfiles/images/2023/05/202305091437412490_5652_5655394_3.png!w690x154.jpg[/img][img=,690,136]https://ng1.17img.cn/bbsfiles/images/2023/05/202305091437499210_2633_5655394_3.png!w690x136.jpg[/img]以上分别为负离子和正离子模式下的色谱图,正离子模式使用的色谱柱是[font='Times New Roman'][font=仿宋]美国[/font]Waters[font=仿宋]公司 [/font][/font]BEH C8[font=仿宋]柱([/font][font=Times New Roman]1.7μm[/font][font=仿宋],[/font][font=Times New Roman]2.1×100mm[/font][font=仿宋]),负离子模式用的色谱柱是[font='Times New Roman'][font=仿宋]美国[/font]Waters[font=仿宋]公司 [/font][/font][font='Times New Roman']HSS T3[font=仿宋]柱([/font][font=Times New Roman]1.8μm[/font][font=仿宋],[/font][font=Times New Roman]2.1×100mm[/font][font=仿宋])[/font][/font][/font]
操作条件对于色谱分离有很大影响。 柱长,柱内径:一般讲,柱管增长,可改善分离能力,短则组分馏出的快些;柱内径小分离效果好,柱内径大处理量大,但柱内径过大,将导致担体不能均匀地分布在色谱柱中。分析用柱管一般内径为3-6毫米,柱长为1-4米。 柱温:是一个重要的操作变数,直接影响分离效能和分析速度。选择柱温的根据是混合物的沸点范围,固定液的配比和鉴定器的灵敏度。提高柱温可缩短分析时间;降低柱温可使色谱柱选择性增大,有利于组分的分离和色谱柱稳定性提高,柱寿命延长。一般采用等于或高于数十度于样品的平均沸点的柱温为较合适,对易挥发样用低柱温,不易挥发的样品采用高柱温。 载气流速:载气流速是决定色谱分离的重要原因之一。一般讲流速高色谱峰狭,反之则宽些,但流速过高或过低对分离都有不利的影响。流速要求要平稳,常用的流速范围每分钟在10-100亳升之间。 固定相:固定相是由固体吸附剂或涂有固定液的担体构成。(1)固体吸附剂或担体粗细:一般采用40-60目、60-80目、80-100目。当用同等长度的柱子,颗粒细的分离效率就要比粗的好些。(2)固定液含量:固定液含量对分离效率的影响很大,它与担体的重量比一般用15%-25%。比例过大有损于分离,比例过小会使色谱峰拖尾。 进样:一般讲进样快,进样量小,进样温度高其分离效果好。对进液体样,速度要快,汽化温度要高于样品中高沸点组分的沸点值,一次汽化,保证色谱峰形不致展宽、使柱效高。当进样量在一定限度时,色谱峰的半峰宽是不变的。若进样量过多就会造成色谱柱超载。一般讲柱长增加四倍,样品的许可量增加一倍。对于常规分析,液体进样量为1-20微升;气体进样量为0.1-5毫升。
操作条件对于色谱分离有很大影响。 柱长,柱内径:一般讲,柱管增长,可改善分离能力,短则组分馏出的快些;柱内径小分离效果好,柱内径大处理量大,但柱内径过大,将导致担体不能均匀地分布在色谱柱中。分析用柱管一般内径为3-6毫米,柱长为1-4米。 柱温:是一个重要的操作变数,直接影响分离效能和分析速度。选择柱温的根据是混合物的沸点范围,固定液的配比和鉴定器的灵敏度。提高柱温可缩短分析时间;降低柱温可使色谱柱选择性增大,有利于组分的分离和色谱柱稳定性提高,柱寿命延长。一般采用等于或高于数十度于样品的平均沸点的柱温为较合适,对易挥发样用低柱温,不易挥发的样品采用高柱温。 载气流速:载气流速是决定色谱分离的重要原因之一。一般讲流速高色谱峰狭,反之则宽些,但流速过高或过低对分离都有不利的影响。流速要求要平稳,常用的流速范围每分钟在10-100亳升之间。 固定相:固定相是由固体吸附剂或涂有固定液的担体构成。(1)固体吸附剂或担体粗细:一般采用40-60目、60-80目、80-100目。当用同等长度的柱子,颗粒细的分离效率就要比粗的好些。(2)固定液含量:固定液含量对分离效率的影响很大,它与担体的重量比一般用15%-25%。比例过大有损于分离,比例过小会使色谱峰拖尾。 进样:一般讲进样快,进样量小,进样温度高其分离效果好。对进液体样,速度要快,汽化温度要高于样品中高沸点组分的沸点值,一次汽化,保证色谱峰形不致展宽、使柱效高。当进样量在一定限度时,色谱峰的半峰宽是不变的。若进样量过多就会造成色谱柱超载。一般讲柱长增加四倍,样品的许可量增加一倍。对于常规分析,液体进样量为1-20微升;气体进样量为0.1-5毫升
看到有帖子说反相柱碳载量越大,极性越小,保留越强,分离度越好。事实真的如此吗?发现很多色谱柱碳载量都差别很大,而且不是新出的产品就一定比后出的高。比如inertsil ODS4碳载量仅有11%,比ODS3的15%低的多,但是宣传的分离效果却要好。那么碳载量对分离究竟有何影响,是越大越好么?谢谢高手解答。
在使用制备色谱过程中,发现增大进样量,会导致相邻峰的分离度降低,甚至重叠在一起无法分离。虽然降低进样量可以解决这个问题,但是又会导致制备效率很低。提高水相的比例,又会使峰变宽,峰高变低,也不能达到好的分离效果。所以想请教一下,如何才能维持较大的进样量,又能达到较好的分离度?
液相色谱柱的含碳量对于保留时间是有影响的,对于分离度的影响大吗?
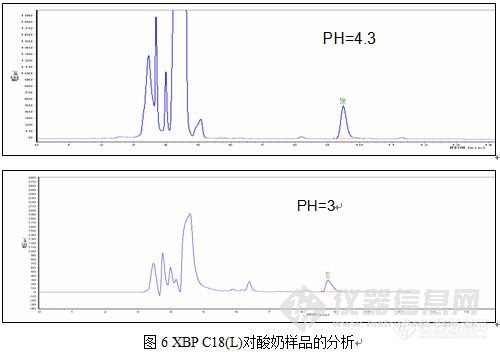
色谱柱表面碳含量和极性对分离的影响一 背景 反相色谱柱如C18是色谱分析工作者在实验中经常使用的工具,但是不同种类的C18色谱柱往往表现不一样。 本研究的目的是从C18色谱柱的表面性质去揭示影响分离效果的因素。主要考虑的表面性质有碳含量和表面极性(硅羟基数)。二 实验原理通过对阿巴斯甜标准品和酸奶样品中阿巴斯甜的测定,对比不同C18色谱柱的分离效果,结合各种色谱柱的表面性质,从而揭示出二者之间的关系。右图为阿斯巴甜的结构:http://ng1.17img.cn/bbsfiles/images/2012/11/201211021113_400878_1871980_3.jpg三 实验部分3.1 样品处理准确称取酸奶5g左右,加入10mL1%三氯乙酸,1mL2%乙酸铅,颠倒数次,6000r/min 离心15min,取上清液于25mL容量瓶,5mLPH=4.3水洗涤沉淀, 6000r/min 离心10min,提取上清液,用PH=4.3水定容。将样品用0.45μm滤膜过滤,将样品进液相检测。http://ng1.17img.cn/bbsfiles/images/2012/11/201211021114_400879_1871980_3.jpg3.2 液相条件 仪器:Agilent HPLC 1200 色谱柱见表1.流动相:甲醇:水=69:31流速:0.8mL/min进样量:20μL检测波长:208nm柱类型长度/mm粒径/μm孔径/Å%C比表面㎡/g封尾封尾类型PHVenusil ASB C184.6*250515012%200否- 0.8-7.5
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]两个峰看上去已经部分重合了,但是分离度却大于1.5,这是算完全分开吗?还有就是数据处理的时候需要扣除空白(溶剂)吗?[/color]
1、分离不完全1。1几个峰重叠,分离不开处理方法:降低载气流速,减少进样量,降低柱温。对于原来能完全分离,使用一段时间后不能完全分离的,表明固定液已经流失,色谱柱的寿命已经完结,需要更换了。1.2分离时间太长使晚馏出的峰扁平。处理方法:可以通过提高柱温来解决。检测器灵敏度太低,使含量少的组分检测不出来。处理方法:可以通过加大进样量,提高检测器灵敏度来解决。2、峰形不对称2.1出现拖尾峰。处理方法:采用强极性的固定液,清除担体活性以及提高柱温来解决。2.2出现平顶形或锯齿形峰。处理方法:通过减少进样量,提高柱温和载气的流速来解决。另外当放大器输入饱和时也会形成平顶峰。
薄层色谱在根据分子量进行分离方面有什么新的进展?
1.系统全面介绍了样品预处理和分析方法;2.本次修订增加了实际样品处理技术、生物样品的沉淀技术、溶剂萃取新技术、微萃取技术等内容;3.适合从事分析检测的初学者阅读.内容简介:全书对样品的预处理和分离方法作了比较系统的讲述,主要内容有分析样品的准备与预处理、沉淀分离技术、萃取分离技术、离子交换分离技术、液相色谱分离技术、电泳分离技术、膜分离技术、泡沫浮选分离技术。此次修订增加了实际样品处理技术、生物样品的沉淀分离技术、溶剂萃取新技术、微萃取技术与加压及旋转薄层色谱分离技术等内容,也对第一版中部分内容作了适当的修订。但由于书中篇幅有限,书中只原则性介绍了相关内容,具体样品的处置还需进一步参考相关文献或技术手册。本书适用于各层次的分析测试工作者,也可供从事其他有关专业的工程技术人员和科研人员参考。目录:第一章 分析样品的准备与预处理/001第一节概述001一、样品采集与处理的基本原则001二、样品制备与处理的注意事项004第二节试样的处理005一、无机样品的处理005二、有机样品的处理009三、生物样品的处理010第三节微波及超声波在样品处理中的应用012一、微波在样品处理中的应用012二、超声波在样品处理中的应用015第四节实际样品处理技术018一、大气样品处理技术018二、水样品处理技术019三、土壤样品处理技术020四、有机及生物样品处理技术021第二章 沉淀分离技术/027第一节沉淀分离技术概述027第二节无机沉淀分离法028一、氢氧化物沉淀分离法028二、硫化物沉淀分离法032三、其他沉淀分离法033第三节有机沉淀分离法033一、生成螯合物的沉淀分离体系034二、生成缔合物的沉淀分离体系036三、生成三元配合物的沉淀分离体系036第四节均相沉淀及共沉淀分离法037一、均相沉淀分离法037二、共沉淀分离法039第五节生物样品的沉淀分离技术043一、等电点沉析044二、盐析沉淀045三、有机溶剂沉析049四、有机聚合物沉析051五、其他沉析技术052第三章 萃取分离技术/055第一节溶剂萃取分离技术055一、溶剂萃取分离基本原理056二、重要的萃取体系060三、有机物的萃取077四、萃取方式与装置079第二节溶剂萃取新技术083一、快速萃取技术083二、反胶团溶剂萃取技术085三、离子液体萃取技术088四、双水相萃取技术090五、微波萃取及超声萃取技术092六、电泳萃取技术097第三节固相萃取技术098一、固相萃取基本原理098二、固相萃取的吸附剂099三、固相萃取装置100四、固相萃取的操作程序100五、固相萃取技术的应用101第四节微萃取技术102一、分散液相微萃取技术102二、分子印迹微萃取技术105三、固相微萃取技术107第五节萃取分离的实际应用110一、应用溶剂萃取分离干扰物质110二、萃取联用分析111三、萃取分离其他示例111第四章 离子交换分离技术/116第一节概述116第二节离子交换剂的结构、性质和分类117一、离子交换剂的结构和性质117二、离子交换树脂的分类与用途120第三节离子交换的基本理论124一、Donnan理论124二、交换反应过程及离子交换选择系数125第四节离子交换的分离操作方法128一、离子交换树脂的选择及预处理128二、离子交换分离操作方法131第五节离子交换分离的实际应用135一、去离子水的制备135二、痕量元素的预富集136三、性质相似离子间的彼此分离137四、生物大分子分离137第五章 液相色谱分离技术/139第一节概述139第二节常压柱色谱分离法140一、吸附柱色谱分离140二、分配柱色谱分离144三、柱色谱分离的操作145第三节平面色谱分离技术146一、纸色谱分离技术146二、薄层色谱分离技术150三、加压及旋转薄层色谱分离技术174第四节柱液相色谱分离技术177一、高效液相色谱分离技术177二、[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分离技术185三、离子对色谱分离技术189四、凝胶色谱分离技术191五、亲和色谱分离技术192六、超临界流体色谱分离技术194第六章 电泳分离技术/197第一节电泳的基本原理197一、电泳迁移率197二、影响迁移率的因素198第二节常用电泳分离技术199一、区带电泳200二、等电聚焦电泳205三、等速电泳206四、毛细管电泳207第三节电泳分析应用210一、在药物分离分析中的应用210二、在生命科学中的应用211三、在临床医学中的应用211四、在环境分析中的应用211五、在作物品种鉴定中的应用212六、在动物和植物科学研究中的应用212第七章 膜分离技术/213第一节概述213第二节膜分离的基本原理214一、反渗透分离法基本原理214二、纳滤分离的基本原理215三、微孔过滤基本原理215四、透析分离基本原理216五、电渗析分离基本原理216六、液膜分离法基本原理217第三节膜材料和膜组件220一、板框式膜组件220二、圆管式膜组件222三、螺旋卷式膜组件223四、中空纤维式膜组件225第四节膜分离技术及应用226一、膜分离的基本流程226二、膜分离的应用227第八章泡沫浮选分离技术/233第一节概述233第二节浮选装置和操作235第三节离子浮选法236第四节沉淀浮选法238一、氢氧化物沉淀浮选238二、有机试剂沉淀浮选239第五节溶剂浮选法240
[align=center][b][size=24px]白酒分析气相色谱仪分离条件的选择[/size][/b][/align][size=18px] 气相色谱仪分析白酒时,除了选择适合的色谱柱和分析方法外,还要选择好分离的蕞佳操作条件,提高色谱柱的分离效能,增大分离度,获得好的分析结果。色谱技术人员根据实际经验总结出白酒分析气相色谱仪分离条件选择,供大家参考。1. 载气及流速、分流比的选择白酒的气相色谱分析,一般使用FID检测器,常用高纯N2做载气,H2做燃烧气,空气作助燃器。若使用一般填充色谱柱,内径在3~4mm,载气的流量在20~100m L/min。对于内径在0.25mm左右的毛细管色谱柱,载气流量在1~2m L/m in。流速太快会降低色谱柱的分离效能,一般高于蕞佳流速10%左右即可,既保证了色谱柱的分离效能,又能获得比较快的分析速度。H2的流速与载气N2流速相当(毛细管色谱柱载气流量+载气分流的流量),实验证明H2流量∶空气流量=1∶10时,FID检测器蕞灵敏。使用毛细管色谱柱时,分流比的选择直接影响到出峰的个数与分离效果。当分流比为30∶1时蕞为恰当,色谱柱分离效能较高,白酒微量成分分离效果好。载气中微量水分、氢气和空气中的微量杂质对色谱柱和检测器影响很大,严重时会使色谱柱失效,基线不稳,噪声增大,检测器灵敏度下降。所以在载气、H2、空气进入色谱仪之前,应当使用分子筛、硅胶等对气体进行净化处理。2. 色谱柱温的选择白酒中的大部分组分沸点都不高,但沸点范围较宽,为了使低沸点的组分有比较好的分离度,一般初始柱温在50℃。程序升温速度不宜过快,否则分离效果变差,程序升温速度太低,出峰时间长,峰形扁平。一般设定在1~8℃/m in,蕞佳程序升温速度在8℃/m in左右,以保证白酒中各组分在相应的温度下得到良好的分离。蕞终温度不能太高,一般不超过250℃,防止色谱柱温过高,引起固定液挥发流失,分离效能变差,出现基线漂移,或导致色谱柱失效。3. 气化室、检测器温度选择白酒的气相色谱分析中,气化室温度一般高于色谱柱温度50~60℃以上,一般控制在120~200℃,以保证进样时白酒试样中所有的组分都能瞬间变成气体。FID检测器的温度通常控制在150~250℃,避免水蒸汽在检测器中凝结,增大噪声而降低检测器的灵敏度,也可以避免出现检测器点火困难的问题。4. 进样量和进样速度的控制使用填充色谱柱时,柱容量比较大,进样量通常在1~5μL,使用10μL或5μL的微量注射器。采用毛细管色谱柱时,柱容量小,进样量通常在0.1~2μL。进样量低不利于使用低含量组分法进行检测,进样量过高则会导致部分组分峰发生重叠,分离不好。进样速度要求比较快,要求1 s内完成,以保证酒样瞬间气化。如果进样速度太慢,就会引起先插进去的针头部分的酒样先气化,导致色谱峰变宽或者异型,峰形不好,分析误差大的问题。每次进样时,应将微量注射器用被测酒样抽洗5次以上并排净气泡,保证待测试样浓度不发生变化,减少进样带来的误差。5. 其他注意事项为了尽可能地减少分析误差,保证分析结果的准确性,要定期老化色谱柱,在高于使用温度20℃,脱开检测器,通以载气10 h以上,让色谱柱中残留的高沸点组分流出,降低仪器噪声,减小高沸点残余物质的干扰。同时还要定期清理色谱柱头和衬管中积累的不挥发物,防止堵塞色谱柱。每进样50次左右就需更换气化室中的硅橡胶垫,保证气化室不漏气,避免出现色谱峰异常现象。在白酒的气相色谱仪分析中,适当地选择分析方法与测定条件,既可以提高色谱分析的分离效能与检测的灵敏度,又可以提高分析结果的准确度。这就需要我们在实际工作中不断探求与创新,找出每种酒样的蕞佳分析条件,做到准确而快速地分析白酒的微量成分,有效地指导白酒的生产、研发和质量监督,保障白酒的食品安全。[/size]
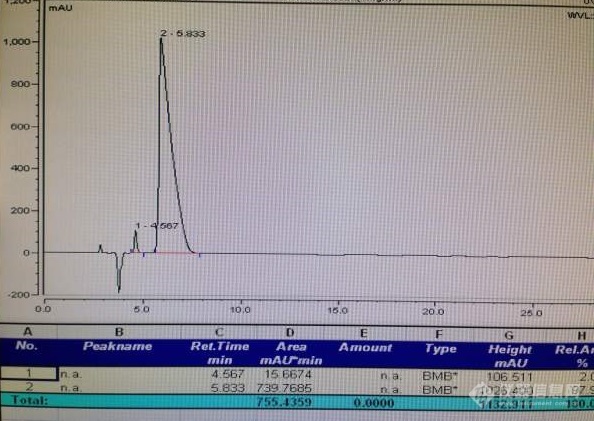
[color=#444444]现想求助一色谱分离问题。[/color][color=#444444] 我做液相色谱时,有两个峰保留时间较近,想让他们分开,大家有什么好方法吗,我已经调过流速、流动相比例和柱温了效果都不明显,具体色谱条件如下,色谱柱:Agilent Eclipse Plus C18(3.5um,4.6*100mm);流动相:0.1%甲酸水:甲醇(95:5),等度;柱温:30℃;流速0.3mL/min;液相色谱图见附件,保留时间为4.567的为图片中的手写结构式,保留时间为5.833的为另一结构式,我想让这两个峰的分离时间达到两分钟以上,因为想让前一个峰进质谱,后一个峰不进,如果浓度降低一起进质谱的话前一个峰就没什么响应了,求高手赐教有什么好办法让这两个峰分的更开一点,谢谢![/color][color=#444444][img=,600,450]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231012429519_6927_1646718_3.jpg!w600x450.jpg[/img][img=,594,421]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231012406386_9342_1646718_3.jpg!w594x421.jpg[/img][/color]
在生物大分子纯化分析特别是蛋白质纯化分析中,色谱是非常重要而且常用的一种技术。 一、凝胶过滤 凝胶过滤又叫分子筛色谱,其原因是凝胶具有网状结构,小分子物质能进入其内部,而大分子物质却被排除在外部。当一混合溶液通过凝胶过滤色谱柱时,溶液中的物质就按不同分子量筛分开了。 二、离子交换色谱 离子交换色谱是在以离子交换剂为固定相,液体为流动相的系统中进行的。离子交换剂是由基质、电荷基团和反离子构成的。离子交换剂与水溶液中离子或离子化合物的反应主要以离子交换方式进行,或借助离子交换剂上电荷基团对溶液中离子或离子化合物的吸附作用进行。 三、吸附色谱 1、 吸附柱色谱 吸附柱色谱是以固体吸附剂为固定相,以有机溶剂或缓冲液为流动相构成柱的一种色谱方法。 2、 薄层色谱 薄层色谱是以涂布于玻板或涤纶片等载体上的基质为固定相,以液体为流动相的一种色谱方法。这种色谱方法是把吸附剂等物质涂布于载体上形成薄层,然后按纸色谱操作进行展层。 3、 聚酰胺薄膜色谱 聚酰胺对极性物质的吸附作用是由于它能和被分离物之间形成氢键。这种氢键的强弱就决定了被分离物与聚酰胺薄膜之间吸附能力的大小。色谱时,展层剂与被分离物在聚酰胺膜表面竞争形成氢键。因此选择适当的展层剂使分离在聚酰胺膜表面发生吸附、解吸附、再吸附、再解吸附的连续过程,就能导致分离物质达到分离目的。 四、 亲和色谱 亲和色谱是根据生物大分子和配体之间的特异性亲和力,将某种配体连接在载体上作为固定相,而对能与配体特异性结合的生物大分子进行分离的一种色谱技术。亲和色谱是分离生物大分子最为有效的色谱技术,分辨率很高。 亲和色谱的原理与众所周知的抗原一抗体、激素一受体和酶一底物等特异性反应的机理相类似,每对反应物之间都有一定的亲和力。正如在酶与底物的反应中,特异的废物(S'')才能和一定的酶(E)结合,产生复合物(E-S'')一样。在亲和色谱中是特异的配体才能和一定的生命大分子之间具有亲和力,并产生复合物。而亲和色谱与酶一底物反应不同的是,前者进行反应时,配体(类似底物)是固相存在;后者进行反应时,底物呈液相存在。实质上亲和色谱是把具有识别能力的配体L(对酶的配体可以是类似底物、抑制剂或辅基等)以共价键的方式固化到含有活化基团的基质M(如活化琼脂糖等)上,制成亲和吸附剂M-L,或者叫做固相载体。而固化后的配体仍保持束缚特异物质的能力。 因此,当把围相载体装人小色谱柱(几毫升到几十毫升床体积)后,让欲分离的样品液通过该柱。这时样品中对配体有亲和力的物质S就可借助静电引力、范德瓦尔力,以及结构互补效应等作用吸附到固相载体上,而无亲和力或非特异吸附的物质则被起始缓冲液洗涤出来,并形成了第一个色谱峰。然后,恰当地改变起始缓冲 液的PH值、或增加离子强度、或加人抑③剂等因子,即可把物质S从固相载体上解离下来,并形成了第M个色谱峰(见图6-2)。显然,通过这一操作程序就可把有效成分与杂质满意地分离开。如果样品液中存在两个以上的物质与固相载体具有亲和力(其大小有差异)时,采用选择性缓冲液进行洗脱,也可以将它们分离开。用过的固相载体经再生处理后,可以重复使用。 上面介绍的亲和色谱法也是特异性配体亲和色谱法。另外还有通用性配体亲和色谱法。这两种亲和色谱法相比,前者的配体一般为复杂的生命大分子物质(如抗体、受体和酶的类似底物等),它具有较强的吸附选择性和较大的结合力。而后者的配体则一般为简单的小分子物质(如金属、染料,以及氨基酸等),它成本低廉、具有较高的吸附容量,通过改善吸附和脱附条件可提高色谱的分辨率。 五、聚焦色谱 聚焦色谱也是一种柱色谱。因此,它和另外的色谱一样,照例具有流动相,其流动相为 多缓冲剂,固定相为多缓冲交换剂。 聚焦色谱原理可以尝试从PH梯度溶液的形成、蛋白质的行为和聚焦效应三方面来阐述。 1、PH梯度溶液的形成 在离子交换色谱中,PH梯度溶液的形成是靠梯度混合仪实现的。例如,当使用阴离子 剂进行色谱时,制备PH由高到低呈线性变化的梯度溶液的方法是,在梯度仪的混合室(这色谱柱者)中装高PH溶液,而在另一室装低PH极限溶液,然后打开色谱柱的下端出口,让洗脱液连续不断地流过柱体。这时从柱的上部到下部溶液的PH值是由高到低变化的。而在聚焦色谱中,当洗脱液流进多缓冲交换剂时,由于交换剂带具有缓冲能力的电荷基团,故PH梯度溶液可以自动形成。 2.蛋白质的行为 蛋白质所带电荷取决于它的等电点(PI)和色谱柱中的PH值。当柱中的PH低于蛋白质的PI时,蛋白质带正电荷,且不与阴离于交换剂结合。而随着洗脱剂向前移动,固定相中的PH值是随着淋洗时间延长而变化的。当蛋白质移动至环境PH高于其PI时,蛋白质由带正电行变为带负电荷,并与阴离子交换剂结合。由于洗脱剂的通过,蛋白质周围的环境PH 再次低于PI时,它又带正电荷,并从交换剂解吸下来。随着洗脱液向柱底的迁移,上述过程将反复进行,于是各种蛋白质就在各自的等电点被洗下来,从而达到了分离的目的。 不同蛋白质具有不同的等电点,它们在被离子交换剂结合以前,移动之距离是不同的,洗脱出来的先后次序是按等电点排列的。
各位有色谱方法开发经验的前辈:现有两个问题需请教1.对于高浓度主峰,峰宽很宽,有6分钟样子,在紧随其后有1杂质峰,与主峰分离度只有0.8,色谱条件除了色谱柱可以更换外,其他条件均不能改变,那么用何种色谱柱可以提高两者的分离度为1.5以上;2.哪类色谱柱更适合于分析高浓度化合物;谢谢各位赐教,不胜感激!
根据所需采集的原始样品和样品基体的性质、所要获得的信息(分析测试的目的)、允许的分析时间和色谱仪器对所分析样品的要求等,决定样品的采集和制备方法及其程序。某些样品(或者粗产品)由于浓度含量较高,可以直接取其一部分母体物质,直接进行色谱分析即可获得满意的结果,而无需预先进行欲测组分的分离和浓缩。但是,大部分的原始样品不适合于色谱仪器直接分析测定,需要对原始样品进行一些处理,制备成适合于色谱仪器分析的样品。很明显,用色谱仪器对这些通过一系列处理后制备的样品进行定性和定量分析,其结果的可靠性和准确性将取决于这些处理过程是否会将欲测组分丢失或使欲测组分发生一些不可预测的变化。满意的定性和定量结果,需要严格而周密的分离和富集方法,如果样品处理过分粗糙,可能会导致样品中欲测组分质的变化,同样会影响定量的误差,使色谱分析的定性和定量结果不准确。另外,某些样品中的某种组分会对色谱分析样品中共存的其他欲测组分产生测定干扰,这样就必须预先分离和除去干扰物,才能完成准确的色谱测定。在许多情况下,可以提供给分析人员的样品量(体积)很少(或者很有限),而且,样品中欲测组分具有较大的不稳定性(或者化学活性),需要经过样品制备才能获得可靠的测定结果,因此,选择和制定周密的样品处理程序和完成准确无误的操作是非常重要的。 由于采用色谱分析的目的不同,诸如痕量分析、物质组成的定性分析、多组分体系中的选择分析、纯度分析、定位分析和结构分析等,使用的样品制备方法和技术也不相同。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]通常采用的样品处理技术有气体萃取、溶剂萃取、固相萃取、超临界萃取、衍生化、膜分离、蒸馏、吸附等技术:液相色谱通常采用的样品处理技术有溶剂萃取、固相萃取、衍生化、微透析、蒸馏等技术。 在实际分析之前,采样和样品处理方法决定分析结果的质量,不合适的或非专业的采样会使可靠正确的测定方法得出错误的结果。目前,在进行色谱分析之前需要进行样品的预浓缩是非常普遍的现象。因为普通色谱方法不可能测定出这样低浓度(10-8 ~10-12,即低于1ng/ ml)的物质。例如:人类对物质气味的感觉阀值都非常低,虽然可以嗅出样品的某些气味,但是色谱的方法却不能将这些气味直接测定出来,必须首先对样品进行预浓缩才能测定出它们。 采集样品涉及从整体中分离出具有代表性的部分进行收集。采样之前,应当对采样的环境和现场进行充分的调査,通常需要弄清楚下面这些问题:(1) 样品中可能会存在的物质组成是什么,它们的浓度水平如何?(2) 样品中的主要组分是什么?(3) 采集样品的地点和现场条件如何?(4) 应该采用非破坏性采样方法还是采用破坏性采样方法?(5) 采样完成后会得到哪些色谱分析的结果? 此外,采集的样品量与使用分析技术的灵敏度成反比。关于采样地点和采样时间应当注意如下的问题:(1) 确定采集样品的最佳时机:(2) 确定采样的位置和采集样品的装置:(3) 采样过程可以保证多长的有效时间:(4) 确定采集样品的间隔时间。 在均匀体系中采集样品可能出现的问题较少,但是,当采集像气溶胶、微粒、泡沫、乳剂、湿的淤渣、溶胶悬浮物等多相性样品时,常常会遇到许多问题。当希望测定的物质及其浓度会随时间变化时,必须仔细研究现场状况以决定采集样品的时机和时间。 目前,有许多的采样和样品制备方法和技术可供选择,诸如美国推荐采用的EPA、OSHA、SDWA、ASTM等方法中都有天然水体样品、空气样品、土壤样品和各种材料样品的样品采集和制备方法供参考。应当根据被采集样品体系的特性和分析测定的目的,选择合适的采样和样品制备技术。无论如何,样品的收集和处理方法及其技术必须遵循下面的原则:(1) 收集的样品必须具有代表性:(2) 采样方法必须与分析目的保持一致,并且采集到你想要的样品:(3) 分析样品制备过程中尽可能防止和避免欲测定组分发生化学变化或者丢失:(4) 在样品处理过程中,如果将欲测定组分进行化学反应时(例如:将不能汽化的欲测定组分转化成可汽化物质的衍生化过程,或者将不适合测定的组分通过化学反应转化成适合测定的物质)这一变化必须是已知的和定量地完成:(5) 在分析样品制备过程中,要防止和避免欲测定组分的玷污,尽可能减少无关化合物引入制备过程:(6) 样品的处理过程应当尽可能简单易行,所用样品处理装置尺寸应当与处理的样品量相适应。 此外,在实际分析样品之前,某些样品可能会发生变化(例如光化学过程、微生物和空气中的氧所引起的变化),致使被测定物质的浓度发生变化。因此,在采样之后应当尽可能快地进行分析样品的制备和分析,或者使用合适的方法消除这种千扰(不使这些变化发生),做好样品的保存。 分析结果的质量与获得的样品代表性密切相关。获得的分析结果只有在样品与监测项目的目标相符时才会有意义。为了对分析结果进行令人满意的评价,应该说明采样点、采样技术、采样频率以及所采集的样品量。在采样前及采样后,采样容器和分析样品之前的样品贮存、运输过程中都必须避免使样品发生改变。环境中的样品通常是非均质的。为了得到有意义的数据,必须对大量样品进行分析。为了保证分析测定结果的可靠性,必须如实地记录采样过程及所采集样品的数量。分析所需样品的总数与所希望得到的信息种类要求有关,若需要样品构成平均值,就必须随机地选择大量样品,将其进行混合、均匀化以获得一个混合样品,从中再取样分析。若需要的是样品组分分布图,就应当对限度样品进行逐个重复分析。 总之,应当根据分析测定的目的、分析测定的对象及其状况、所具备的分析测定的条件,选择并制定最佳的可实施的色谱分析样品处理程序。
‘有奖问答’选择题:在色谱的流出曲线上,相邻两组分色谱峰的分离度主要取决于( )A. 分配系数 B. 保留参数 C. 色谱柱温度 D. 载气流速
维权声明:本文为环烯醚萜原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。老生常谈——开放ODS柱色谱在分离纯化中的应用什么是ODS? ODS是英文octadecyl silane的缩写,意思是十八烷基硅烷,是以硅胶为基质键合的C18填料,属于反相色谱。ODS柱色谱,简单地说,就是指用ODS装成的色谱柱啦。“开放”一词,是指未对色谱柱施加任何压力,让其在重力作用下洗脱;相对于开放ODS柱色谱,有中低压柱色谱,高低柱色谱等等。 我们平时所说的反相HPLC,一定程度上来说,其实也可以说是ODS柱色谱,只不过它的分离效果更好,还配备了紫外或者示差等高级地检测手段,其实质还是属于反相柱色谱。适用范围有哪些? 首先需要说明:ODS在分离纯化的过程中,起着极为巨大的作用!! 就化合物极性而言,ODS适合分离极性中等偏大的化合物类型。对于极性较小的化合物,应该考虑用硅胶、凝胶等手段进行分离纯化,绝对不要尝试ODS,唯一的后果只能是:样品全部死吸附,根本不可能洗脱!! 某些行业,前处理一般是萃取,比如环己烷/石油醚、乙酸乙酯/氯仿、正丁醇、水。水层的、正丁醇层的样品,一般都可以用ODS进行分离纯化;乙酸乙酯层的样品,极性偏大的部分可以考虑用ODS;至于环己烷层的样品,千万不要使用ODS,那将是:上样量多少,死吸附就有多少!! 就化合物分类而言,ODS对于黄酮苷类、环烯醚萜苷类、糖苷类等成分都能有一定的分离效果,也有很多成功的分离纯化实例。但是,对于苷元类化合物,则需要慎重考虑,其实还是极性大小的问题:苷元类化合物,一般极性较小。如何装柱、上样、洗脱? 装柱:新买来的ODS,用甲醇浸泡过夜后即可装柱。具体的装柱方法,跟硅胶的装柱子法是一样的,记得用甲醇装柱就行。装完以后,置换成起始流动相系统,即可投入使用了。 上样:分为两种,一种是湿法上样,另一种是拌样。湿法上样,不必多说,就是将样品溶解至一定体积(强烈建议用起始流动相溶解,但体积不可过大,太大相当于原点变大,分离度变差),然后上样;至于拌样,很多的观点表示ODS不应该拌样,我在这里指出,依我个人经验来看,拌样是可以的,尤其对于一些溶解性较差的样品,拌样后分离的效果,比湿法上样好很多,大家可以尝试。(所谓拌样,就是指将你的样品溶解,然后从柱子里面掏出小部分ODS,然后进行拌匀,注意不要过载) 洗脱过程:如果你有ODS薄层板,你可以先点板看看样品大概的极性大小。如果你没有ODS板,你拿到的是“盲样”,一般可以采取常规的梯度洗脱,如水——30%甲醇/水——50%甲醇/水——70%甲醇/水——100%甲醇,然后依据各流分样品量的大小进一步进行细分;经过常规的梯度洗脱以后,假设,你的样品集中在50%的甲醇/水部分,进行细分的时候,你就可以选择40%甲醇/水——45%甲醇/水——50%甲醇/水——100%甲醇这样的梯度。 流动相的选择:其实就是反相流动相,无非就是甲醇/水,乙腈/水,流动相中也可以加入一些其他物质。我曾经很多次加入乙酸,用来改善拖尾的现象(点ODS薄层板分析过)。至于缓冲盐,我没有试过,具体情况不太了解。洗脱样品的如何处理? 洗脱下来的样品,可用硅胶薄层板进行点板分析,然后将相同流分进行合并,也可以采用HPLC检测合并。 我认为,对于一些量小的流分,相差不是很大的,尽量合并,避免样品的分散; 对于一些量大的流分,可合可不合,反正量大,合与不合的效果是一样的(无非就是多占点空间); 从ODS上洗脱的流分,很可能是纯品;易结晶的样品,会在溶剂挥发的过程中结晶析出来,要注意观察;柱子被污染如何处理? 污染是正常的,见过那么多,也用过那么多ODS柱子,没有哪一根能在使用一段时间后保持“洁白无瑕”的。 被污染,一般情况下就是用甲醇冲洗,一直到冲洗液蒸干没有杂质为止,就可以进行下一批样品的分离了。 也许有人问:你这么处理彻底吗?回答是否定的,但是,对于ODS柱色谱,这样的处理已经足够了。 道理很简单,用ODS进行分离的样品,一般极性不会很小,70%甲醇/水基本能全部冲洗下来。现在已经用甲醇冲洗得差不多,在你对下一批样品进行分离的时候,上一批残留的杂质也就只有100%的甲醇能洗脱下来,根本不会影响。 如果一定要处理,可以采用甲醇:异丙醇=1:1冲洗,这种做法,算是终极做法了。
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。 可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可点击头像联系
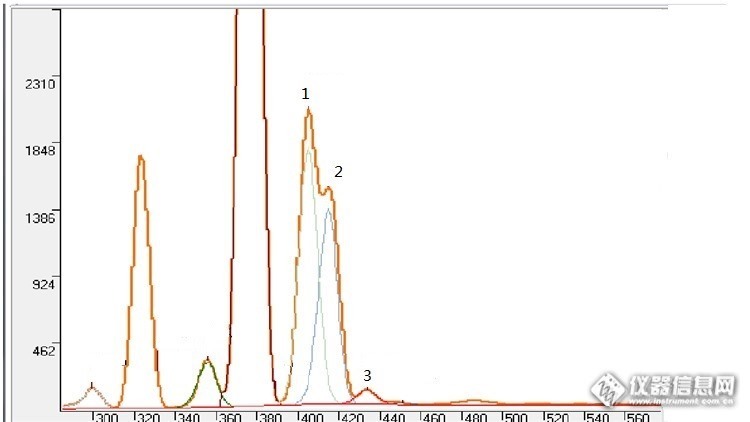
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可联系我 电话:13926563756 qq:648048428 Email: midstone@126.com
色谱条件:0.1%磷酸水,乙腈,30℃,梯度运行,在乙腈达到95%的时候两个物质出峰,且位置相同。合成人员走了联苯甲醚定位和联苯乙酸乙酯定位,发现这两个物质出峰位置相同,因此想分开这两个物质……,联苯乙酸乙酯的沸点较高,300多℃,没办法走[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]……求问可以如何改变条件让他们两个分离?
1 稀释样品对组成复杂的样品, 若待测离子对树脂亲合力相差颇大, 就要作几次进样, 并用不同浓度或强度的淋洗液或梯度淋洗. 对固定相亲合力差异较大的离子, 增加分离度的最简单方法是稀释样品或作样品前处理. 例如盐水中SO2 -4 和Cl - 的分离. 若直接进样, 其色谱峰很宽而且拖尾, 表明进样量已超过分离柱容量, 在常用的分析阴离子的色谱条件下, 30min 之后Cl - 的洗脱仍在继续. 在这种情况下,在未恢复稳定基线之前不能再进样. 若将样品稀释10 倍之后再进样就可得到Cl - 与痕量SO2 -4 之间的较好分离. 对阴离子分析推荐的最大进样量, 一般为柱容量的30 % , 超过这个范围就会出现大的平头峰或肩峰2 改变分离和检测方式若待测离子对固定相亲合力相近或相同, 样品稀释的效果常不令人满意. 对这种情况, 除了选择适当的流动相之外, 还应考虑选择适当的分离方式和检测方式. 例如, NO-3 和ClO-3 , 由于它们的电荷数和离子半径相似, 在阴离子交换分离柱上共淋洗. 但ClO-3 的疏水性大于NO-3 , 在离子对色谱柱上就很容易分开. 又如NO-2 与Cl - 在阴离子交换分离柱上的保留时间相近, 常见样品中Cl - 的浓度又远大于NO-2 , 使分离更加困难, 但NO-2 有强的UV 吸收, 而Cl - 则很弱, 因此, 应改用紫外作检测器测定NO-2 , 用电导检测Cl - , 或将两种检测器串联, 于一次进样同时检测Cl - 与NO-2 . 对高浓度强酸中有机酸的分析, 若采用离子排斥, 由于强酸不被保留, 在死体积排除, 将不干扰有机酸的分离.3 样品前处理对高浓度基体中痕量离子的测定, 例如海水中阴离子的测定, 最好的方法是对样品作适当的前处理. 除去过量Cl - 的前处理方法有: 使样品通过Ag+ 型前处理柱除去Cl - , 或进样前加AgNO3 到样品中沉淀Cl - 也可用阀切换技术, 其方法是使样品中弱保留的组分和90 %以上的Cl - 进入废液, 只让10 %左右的Cl - 和保留时间大于Cl - 的组分进入分离柱进行分离. 对含有大的有机分子的样品, 应于进样前除去有机物, 较简单的方法是用Dionex 的前处理柱OnGuard 的RP 或P 柱或在线阀切换除去有机基体.4 选择适当的淋洗液[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分离是基于淋洗离子和样品离子之间对树脂有效交换容量的竞争, 为了得到有效的竞争, 样品离子和淋洗离子应有相近的亲合力. 下面举例说明选择淋洗液的一般原则. 用CO2 -3 2HCO-作淋洗液时, 在Cl - 之前洗脱的离子是弱保留离子, 包括一价无机阴离子、短碳链一元羧酸和一些弱离解的组分, 如F- , 甲酸, AsO-2 , CN- 和S2 - 等. 对乙酸, 甲酸与F- 、Cl - 等的分离应选用较弱的淋洗离子, 常用的弱淋洗离子有HCO-3 , OH- 和B4O2 -7 . 由于HCO-3 和OH- 易吸收空气中CO2 , CO2 在碱性溶液中会转变成CO2 -3 , CO2 -3 之淋洗强度较HCO-3 和OH- 大, 因而不利于上述弱保留离子的分离. B4O2 -7 亦为弱淋洗离子, 但溶液稳定, 是分离弱保留离子的推荐淋洗液. 中等强度的碳酸盐淋洗液对高亲和力组分的洗脱效率低.对离子交换树脂亲合力强的离子有两种情况, 一种是离子的电荷数大, 如PO3 -4 , AsO3 -4 和多聚磷酸盐等 一种是离子半径较大, 疏水性强, 如I - , SCN- , S2O2 -3 , 苯甲酸和柠檬酸等. 对前者以增加淋洗液的浓度或选择强的淋洗离子为主. 对后一种情况, 推荐的方法是在淋洗液中加入有机改进剂(如甲醇、乙腈和对氰酚等) 或选用亲水性的柱子, 有机改进剂的作用主要是减少样品离子与离子交换树脂之间的非离子交换作用, 占据树脂的疏水性位置, 减少疏水性离子在树脂上的吸附, 从而缩短保留时间, 减少峰的拖尾, 并增加测定灵敏度.在[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]中, 可由加入不同的淋洗液添加剂来改善选择性, 这种淋洗液添加剂只影响树脂和所测离子之间的相互作用, 而不影响离子交换. 对与树脂亲合力较强的离子, 如一些可极化的离子, I-和ClO4- , 以及疏水性的离子, 苯甲酸和三乙胺等, 在淋洗液中加入适量极性的有机溶剂如甲醇或乙腈, 可缩短这些组分的保留时间并改善峰形的不对称性. 为了减少样品离子与树脂之间的非离子交换作用, 减少树脂对疏水性离子的吸附, 在阴离子分析中, 可在淋洗液中加入对氰酚. 如测定1 %NaCl中的痕量I- 和SCN- 时, 加入对氰酚占据树脂对I- 和SCN- 的吸附位置, 从而减少峰的拖尾并增加测定灵敏度. [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]IC[/color][/url] 中, 一价淋洗离子洗脱一价待测离子, 二价淋洗离子洗脱二价待测离子, 淋洗液浓度的改变对二价和多价待测离子保留时间的影响大于一价待测离子. 若多价离子的保留时间太长, 增加淋洗液的浓度是较好的方法.
接触色谱的时间有两年了,绝大多数实验都按照已有的常规进行分析(有些已经形成相应的标准),另外一小部分样品中含有的物质大部分已经能确定,只需要根据已知的化学物质特性进行相应的分析,所以分析难度是大大降低了。虽然如此,我还是仔细思考了以往的实验的过程,回忆当时条件选择的原因,写出这段文字,希望大家不吝赐教。1、样品处理我常用的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测器是FID,要求样品中不能含有水分。对于微量的水分,要加入无水硫酸钠并搅拌,待无水硫酸钠均结晶成粉状结晶体表示已除去上清液中的水分。对含水量较多的样品,要进行萃取后才能进行色谱分析。2、色谱柱选择a、色谱柱极性通常情况下,根据被分析物质的物化性质及相似相溶原理,选择合适的柱子。现阶段最常用的是毛细管色谱柱。选择与被测物相近极性的固定液通常有效,但不一定是最佳的分离固定相。但在分离同等情况下,最好选择低极性的固定液。原因是低极性的柱子有较高柱效,并且有较高的抗氧化能力及较高的使用温度。在一般情况下,可以使用一些常用柱子进行试验,比如SE-30。如果效果不太理想,再按上述规则按极性由高到低的顺序选择色谱柱。b、色谱柱直径内径越小,柱效越高。内径大,允许进样量就越大。c、色谱柱柱长一般使用30m的柱子,当10-15m柱子能满足分离要求时,尽量使用短柱子,可以减少分离时间。50-60m长的柱子适合分离含有多各组分的混合物,分析时间也相应增长。d、色谱柱膜厚薄液膜常用于分析含量较高的组分,而厚液膜常用于痕量组分分析。3、汽化温度进样口温度应高于样品中含量最大的组分的沸点20℃左右,以保证样品快速汽化,但温度过高又可能使样品分解,所以对于未知的样品要将进样口温度设置高一些进行试验。4、柱温柱温应接近样品中含量最大的组分的沸点,开始试验初始温度可以用恒温,运行时间长些,然后根据实验结果调整柱温,在适当的位置加入程序升温,减少分析时间。5、检测器温度对于FID检测器,一般250-300℃即可满足分离要求。对于特殊样品,可适度调整。6、分流比不分流的情况适用于痕量分析和沸程较宽的样品分析。分流进样用于大部分可挥发的样品,只在灵敏度太低的情况下才考虑不分流进样和其它进样方式。7、载气流速载气流速过高或过低都影响分离效果。氮气做载气时,流速一般为20-70mL/min。8、进样量因衬管体积有限,分流进样的进样量不超过2微升,具体进样量可根据样品的浓度调节。
1、稀释样品 对组成复杂的样品,若待测离子对树脂亲合力相差颇大,就要作几次进样,并用不同浓度或强度的淋洗液或梯度淋洗。对固定相亲合力差异较大的离子,增加分离度的最简单方法是稀释样品或作样品前处理。例如盐水中SO2-4和Cl-的分离。若直接进样,其色谱峰很宽而且拖尾,表明进样量已超过分离柱容量,在常用的分析阴离子的色谱条件下,30min之后Cl-的洗脱仍在继续。在这种情况下,在未恢复稳定基线之前不能再进样。若将样品稀释10倍之后再进样就可得到Cl-与痕量SO2-4之间的较好分离。对阴离子分析推荐的最大进样量,一般为柱容量的30%,超过这个范围就会出现大的平头峰或肩峰 2、改变分离和检测方式 若待测离子对固定相亲合力相近或相同,样品稀释的效果常不令人满意。对这种情况,除了选择适当的流动相之外,还应考虑选择适当的分离方式和检测方式。例如,NO-3和ClO-3,由于它们的电荷数和离子半径相似,在阴离子交换分离柱上共淋洗。但ClO-3的疏水性大于NO-3,在离子对色谱柱上就很容易分开。又如NO-2与Cl-在阴离子交换分离柱上的保留时间相近,常见样品中Cl-的浓度又远大于NO-2,使分离更加困难,但NO-2有强的UV吸收,而Cl-则很弱,因此,应改用紫外作检测器测定NO-2,用电导检测Cl-,或将两种检测器串联,于一次进样同时检测Cl-与NO-2。对高浓度强酸中有机酸的分析,若采用离子排斥,由于强酸不被保留,在死体积排除,将不干扰有机酸的分离。 3、样品前处理 对高浓度基体中痕量离子的测定,例如海水中阴离子的测定,最好的方法是对样品作适当的前处理。除去过量Cl-的前处理方法有:使样品通过Ag+型前处理柱除去Cl-,或进样前加AgNO3到样品中沉淀Cl-;也可用阀切换技术,其方法是使样品中弱保留的组分和90%以上的Cl-进入废液,只让10%左右的Cl-和保留时间大于Cl-的组分进入分离柱进行分离。对含有大的有机分子的样品,应于进样前除去有机物,较简单的方法是用Dionex的前处理柱OnGuard的RP或P柱或在线阀切换除去有机基体。 4、选择适当的淋洗液 离子色谱分离是基于淋洗离子和样品离子之间对树脂有效交换容量的竞争,为了得到有效的竞争,样品离子和淋洗离子应有相近的亲合力。下面举例说明选择淋洗液的一般原则。用CO2-32HCO-作淋洗液时,在Cl-之前洗脱的离子是弱保留离子,包括一价无机阴离子、短碳链一元羧酸和一些弱离解的组分,如F-,甲酸,AsO-2,CN-和S2-等。对乙酸,甲酸与F-、Cl-等的分离应选用较弱的淋洗离子,常用的弱淋洗离子有HCO-3,OH-和B4O2-7。由于HCO-3和OH-易吸收空气中CO2,CO2在碱性溶液中会转变成CO2-3,CO2-3之淋洗强度较HCO-3和OH-大,因而不利于上述弱保留离子的分离。B4O2-7亦为弱淋洗离子,但溶液稳定,是分离弱保留离子的推荐淋洗液。中等强度的碳酸盐淋洗液对高亲和力组分的洗脱效率低。 对离子交换树脂亲合力强的离子有两种情况,一种是离子的电荷数大,如PO3-4,AsO3-4和多聚磷酸盐等;一种是离子半径较大,疏水性强,如I-,SCN-,S2O2-3,苯甲酸和柠檬酸等。对前者以增加淋洗液的浓度或选择强的淋洗离子为主。对后一种情况,推荐的方法是在淋洗液中加入有机改进剂(如甲醇、乙腈和对氰酚等)或选用亲水性的柱子,有机改进剂的作用主要是减少样品离子与离子交换树脂之间的非离子交换作用,占据树脂的疏水性位置,减少疏水性离子在树脂上的吸附,从而缩短保留时间,减少峰的拖尾,并增加测定灵敏度。 在离子色谱中,可由加入不同的淋洗液添加剂来改善选择性,这种淋洗液添加剂只影响树脂和所测离子之间的相互作用,而不影响离子交换。对与树脂亲合力较强的离子,如一些可极化的离子,I-和ClO4-,以及疏水性的离子,苯甲酸和三乙胺等,在淋洗液中加入适量极性的有机溶剂如甲醇或乙腈,可缩短这些组分的保留时间并改善峰形的不对称性。为了减少样品离子与树脂之间的非离子交换作用,减少树脂对疏水性离子的吸附,在阴离子分析中,可在淋洗液中加入对氰酚。如测定1%NaCl中的痕量I-和SCN-时,加入对氰酚占据树脂对I-和SCN-的吸附位置,从而减少峰的拖尾并增加测定灵敏度。IC中,一价淋洗离子洗脱一价待测离子,二价淋洗离子洗脱二价待测离子,淋洗液浓度的改变对二价和多价待测离子保留时间的影响大于一价待测离子。若多价离子的保留时间太长,增加淋洗液的浓度是较好的方法。
目的:建立一个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件同时分离测定环孢素A中乙醇及丙二醇的含量。方法:以GDX-101为固定相,柱长为2 m,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标。结果:乙醇及丙二醇进样量分别在2.0~6.0 μg,1.0~3.0 μg,其峰面积与浓度呈良好的线性关系,加样回收率分别为99.9%(RSD<0.8%,n=5),101.4%(RSD<1.1%,n=5),精密度良好。结论:此[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件可同时测定环孢素A中乙醇及丙二醇的含量,方法简便准确。关键词 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 乙醇 丙二醇 环孢素A山地明(环孢素A)为诺华制药有限公司的产品,是一种免疫抑制剂,用于器官移植和骨髓移植中的抑制排斥现象以及自身免疫疾病。厂方质量标准中乙醇及丙二醇的含量采用石英毛细管柱测定,此种色谱柱在国内使用不普及,我们经多次试验,摸索出一较好的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,适用于国内检测,即以GDX-101为固定相,柱长为2 m,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标,可同时分离测定环孢素A中乙醇及丙二醇的含量,改进后的方法,乙醇与正丙醇的分离度为3.1,丙二醇与正丙醇的分离度为5.0,符合中国药典1995年版中乙醇量度检查的分离度要求[1],操作简便,结果准确可靠。1 仪器与试药 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:SP-6890 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱:玻璃柱,长2 m,固定相为GDX-101。 乙醇、异丙醇、丙二醇均为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]纯,二甲基亚砜为色谱纯。 样品:环孢素A胶囊(山地明),由诺华公司提供,批号为187MFD0797;241MFD0797;166MFD0797;483MFD0797;477MFD0797。 标准贮备液及内标贮备液:精密称取[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]级的乙醇及丙二醇2.50及1.25 g分置50 mL容量瓶中,加二甲基亚砜至刻度,摇匀,作为标准贮备液;精密量取正丙醇5.0 mL置50 mL量瓶中,加二甲基亚砜至刻度,摇匀,作为内标贮备液。2 试验方法与结果2.1 色谱条件 采用GDX-101为固定相,柱长为2 m,氮气为载气,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,即初始为165 ℃,保持12 min,以40 ℃。min-1升至280 ℃,并保持20 min,检测器温度为280 ℃,进样量为2 μL。2.2 分离度试验 称取乙醇、丙二醇及正丙醇各50 mg置同一50 mL量瓶中,加二甲基亚砜至刻度,摇匀,进样2 μL,按上述色谱条件试验,记录色谱图,见图1-A,乙醇、丙二醇及正丙醇的保留时间分别为1.15,2.22,7.54 min,计算乙醇与正丙醇及丙二醇与正丙醇的分离度,其分离度分别为3.1和5.0。图1 分离度色谱(A)及样品测定(B)色谱图1.乙醇 2.正丙醇 3.丙二醇 4.二甲基亚砜2.3 线性范围及标准曲线 分别精密量取乙醇和丙二醇标准贮备液1.0,1.5,2.0,2.5,3.0 mL,分别置50 mL量瓶中,并分别加入内标贮备液1.0 mL,使乙醇终浓度为1.0,1.5,2.0,2.5,3.0 mg.mL-1,丙二醇的终浓度为0.5,0.75,1.0,1.25,1.5 mg.mL-1,分别进样2 μL,以乙醇及丙二醇的进样量为横坐标,以它们的峰面积与内标峰面积之比为纵坐标,分别进行线性回归,结果线性关系良好,乙醇、丙二醇回归方程分别为:A=8.935×103C+7.858×102 r=0.998 8A=8.086×103C-1.649×102 r=0.999 92.4 精密度试验 用乙醇与丙二醇浓度分别2.0及1.0 mg.mL-1的溶液,重复进样5次,结果乙醇与丙二醇的RSD分别为0.7%和1.0%,精密度良好。2.5 回收率试验 采用加样回收法,取已知乙醇与丙二醇含量的样品2粒,用二甲基亚砜溶解,置50 mL量瓶中,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,精密量取此溶液4.0,4.5,5.0,5.5,6.0 mL,分别加入乙醇与丙二醇的浓度分别为2.0 mg.mL-1及1.0 mg.mL-1的标准溶液6.0,5.5,5.0,4.5,4.0 mL,混匀,量取混匀后的溶液2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定这5份溶液的乙醇和丙二醇含量,计算回收率,乙醇的平均回收率为99.9%(RSD<0.8%,n=5),丙二醇的平均回收率为101.4%(RSD<1.1%,n=5)。2.6 样品的测定 取乙醇和丙二醇标准贮备液2.0 mL,内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为对照品溶液;取环孢素A胶囊2粒,置50 mL量瓶中,用二甲基亚砜溶解,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为样品溶液;分别量取对照品溶液和样品溶液各2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],按上述色谱条件测定,以内标法计算含量,即得;见图1-B。2.7 对比试验结果 取环孢素A样品5批,用改进后的方法测定样品中乙醇和丙二醇的含量,与厂方测定数据相比,结果基本吻合,见表1。表1 乙醇和丙二醇对比试验结果(%) 批号 本法结果 厂方测定数据 乙醇 丙二醇 乙醇 丙二醇 187MFD0797 101.0 106.3 100.5 105.0 241MFD0797 99.2 99.2 100.6 100.6 166MFD0797 101.7 102.7 101.3 103.0 483MFD0797 98.8 96.8 99.3 97.2 477MFD0797 99.1 98.1 98.9 97.7 3 讨论3.1 本法与原厂方方法相比,方法更为简便,条件普及,有利于对样品质量的控制。3.2 原厂方标准在测定乙醇含量时,以正丁醇为溶剂,由于正丁醇的保留时间与丙二醇过于接近,分离度达不到要求,本法采用二甲基亚砜为溶剂,不影响样品的溶解,同时使丙二醇与二甲基亚砜的分离度符合定量分析的要求。3.3 曾用固定相为GDX-401的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]柱进行检测,乙醇与正丙醇得到完全分离,但丙二醇与溶剂峰重叠,分离度达不到要求。3.4 采用程序升温,可使溶剂出峰时间加快,缩短分析时间。王俊秋(北京市药品检验所 北京 100035)庞青云(北京市药品检验所 北京 100035)余立(北京市药品检验所 北京 100035)参考文献1,中国药典.1995.二部:附录44
拥有良好的分离度是得到准确的定性定量结果的重要保障,在进行色谱分析时,如果发现分离度下降,应如何处理?
液相色谱中死时间几种测定方法1:有响应的溶剂出峰时间为死时间。2:进样后的阀切换峰,对应的时间为死时间。3:工作站中输入色谱柱的空余体积或孔隙率,自动计算。(对于C18柱,也可以用硝酸钠或硫脲等在色谱柱上完全不保留组分的出峰时间来测定死时间。)影响分离度的因素有三个因素控制两个色谱峰之间的分离度——容量因子,选择性,柱效容量因子反映样品分子和固定相及流动相之间的作用力,选择性是说明色谱系统区分两个或多个色谱峰的能力,柱效与色谱峰的宽度有关,很明显要达到一定的分离度,宽色谱峰要比窄色谱峰需要更大的分离度选择性。理论塔板数越高柱效越高,柱效的高低受柱内效应和柱外效应的影响。选择性是固定相区分两个被分离样品组分的能力,用容量因子之比进行计算,它是两个被分离色谱峰顶点距离的量度,如果选择性是Ⅰ,则两个组分完全不能分离。选择性数值越高,分离越好。由于选择性取决于被分离物的物理和化学结构,流动相和固定相,流动相组成,PH,色谱柱温度,流动相添加剂,因此,尽量优化实验条件提高选择性以降低成本。容量因子为物质的特性,当分析条件一定时,容量因子为固定值。溶剂的洗脱强度与其极性有关:反相色谱:溶剂的极性越强,洗脱强度越弱。正相色谱:溶剂的极性越强,洗脱能力越强。(注意:不可以使用纯水作为正想色谱的流动相)一般样品分析要求:容量因子大于2小于5
谈色谱仪用数据处理装置-1 我们知道由于色谱法本身的特点,决定了它的分析数据后处理过程的复杂性,所以分析结果的可用性很大程度上与数据处理装置的可靠性和数据处理的好坏有关。过去常说,色谱仪的设计制造心脏部分是检测器,色谱分析工作者使用操作仪器心脏是选择一根好的色谱柱。现在是否还应强调要获得准确可靠的定性定量结果,另一心脏部分是数据处理装置的功能优劣和正确使用。目前使用的色谱数据处理装置(如色谱数据微处理机,色谱数据处理工作站等)已不是色谱分析发展初期使用的记录仪——仅仅提供一张测量分析组分的保留时间,峰面积的色谱图。可以说那时的数据处理对色谱检测器工作状态和样品组分得分离没有什么贡献。而现代色谱数据处理已不是简单的作为数据处理,而是可以对色谱仪性能(如通过计算机软件降低噪声提高信噪比)和分离(如未分离的峰拟合数据处理)能做出巨大贡献。换句话说,当色谱仪性能欠佳和建立的色谱分析方法使某些组分分离不理想时,可以选择性能比较好的色谱数据处理装置(通过设置合理的一套参数)给以补偿。或者说,再好的色谱仪和分析方法如果没有选择好一台合适的色谱数据处理装置,是不会获得准确可靠的定性定量结果的。综上所述,人们对色谱数据处理装置的重要性应有更加深入认识的必要。本文编写的目的就是对那些接触色谱仪,从事色谱分析时间不长的同行,搞一个讨论色谱数据处理装置:工作原理;功能选择;使用维护等的机会。为了通俗,简捷,上述所涉及的内容用问答的方式加以讨论。在这里还应特别指出的有三点:1. 随着样品分析种类越来越复杂,含量越来越低,分析速度越来越快,对仪器的数据处理装置要求也越来越高,再加上微电子技术,计算机技术大力发展和有关软件开发,色谱数据处理装置和技术仍是色谱仪制造与应用中十分活跃的领域之一。因此,在介绍内容上,有些提法和见解难免欠妥,望广大读者或同仁指正和踊跃参加讨论。2. 在色谱图中当遇到峰型对称性差,重叠峰的峰高比相差较大,尖峰时噪声较严重时等,色谱样品分析的最终分离效果的判决权和定量精度准确度掌握在色谱数据处理的操作者手中,仅从这一点看毫不夸张地说,目前应是加强对色谱分析工作者重视的时候了。3. 对于不同操作者,使用不同类型数据处理,定量分析结果的重现性如何考察,目前国内外恐拍还没有什么好办法,这一点已引起美国食品与药品管理部门的不安与重视。最好的解决办法是建立一种标准方法和装置能针对各种色谱数据处理装置(系统),能进行客观正确的评价。这个问题亦是国内有关专家经常提出和考虑的问题,要解决好这个问题还需多方协同努力,当然越快越好。问-1:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法输出电信号的特点是什么?[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]输出的电信号有三个主要特点:1.目前用于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的几种常用检测器(TCD,FID,ECD,FPD,NPD 等)不能依据被分析组分的分子结构转变输出特微电信号,这点和质谱,光谱有很大区别;2.和某些分析方法相比色谱分析法使用的是相对定量方法;3.[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]输出信号快速(再某些情况下用毫/秒计算);信号小(最小为≤1×10-14A);为模拟电信号,鉴于以上原因,色谱仪输出电信号无法用简单的方法进行定性定量,而必须先把模拟电信号用记录仪记录下来或把模拟信号转换成数字信号贮存下来,然后根据不同分析要求,再做处理,以获得有关被分析组分的定性定量结果或其他信息。不难看出由于色谱法分析本身特点决定了它的数据后处理过程的复杂性,所以分析结果的可用性很大程度上与数据处理好坏有关。问-2:什么是信息?什麽是数据?英文信息(information)可以翻译成情报一般认为是数据,消息中所包含的意义。它不随载荷的物理设备形式而改变,我们也可定为人或机器提供的关于现时世界事实的知识。数据是表示信息的,它定义为载荷信息的物理符号。信息与数据二者在概念上即不可分离又有一定的区别。例如:有一种物质含量为99.999%,这一数据表示这种物质的纯度相当高的这一信息。不难看出,数据是信息的具体表现,是人为的。因为我们也可以把含量为99.999%的物质叫做色谱纯物质或高纯物质等.问-3: 什么是数据处理? 什麽是分析仪的数据处理?数据处理是指将数据转换成信息的过程,它包括对数据的收集(采集),存储,传送,检索,分类,加工计算,输出(报告)等一系列活动.其基本目的是从大量的杂乱无章的难以理解的数据中整理出对人们有价值,有意义的信息作为决策的依据。现代分析仪器分析结果,是一些复杂的由数据组成的一系列谱图如色谱,质谱, 光谱,核磁共振波谱等。这些分析数据必须经过各式各样运算才能得出定性定量结果,具体到色谱分析多指对各种检测器输出信号进行滤波,放大,采集,平滑,存储,判峰,基线校正,确定峰高,保留时间,计算峰面积并进行校正,各种定量计算, 最后输出含有定性定量结果以及其它信息的分析报告的这个过程,称为“色谱 数据处理”。
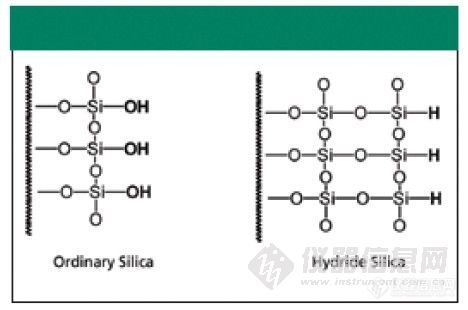
好不容易翻译过来的,如果有错,请大家原谅。两种分离模式的比较:亲水相互作用色谱HILIC 与反反相色谱ANPJoseph Pesek,Maria T. MatyskaDepartment of Chemistry, San Jose State University, San Jose, CaliforniaPlease direct correspondence to Joseph Pesek at pesek@sjsu.eduhttp://ng1.17img.cn/bbsfiles/images/2011/04/201104121803_288425_1604317_3.jpg在文献(1)及其他很多文献中,经常可以看到两种相似的色谱分离机理的色谱柱,一种是亲水相互作用色谱(hyd rophilic interaction liquid chromatography,HILIC),另外一种是水相正相色谱(aqueous normal phase,ANP,也称反反相色谱)。但是,事实上这两种色谱柱在保留模式上是不一样的。 本文旨在对HILIC 和ANP 色谱机理做一个准确的定义并对何时使用哪种色谱柱做一个明确的指导。实验部分实验所使用的色谱柱为UDC Cholesterol(75mm×4.6mm)及Bidentate C18(BD C18,150mm×4.6 mm 或20 mm×2.0 mm)来自于MicroSolv Technology (Eatontown,New Jersey)。流动相中的乙腈来自于B&J(Muskegon,Michigan),水使用来自Milli-Q 仪器(Millipore,Bedford,Massachusetts)。甲酸购自Spectrum Chemical(Gardena,California)。HPLC为安捷伦1050 型,配有自动进样器和二极管阵列检测器(Wilmington,Delaware)。样品浓度范围为0.1-1mg/mL,进样量为1-5μL。反反相实验中的流动相含有0.1%甲酸。结果与讨论HILIC: 亲水色谱是专为保留和分离极性-离子型化合物所设计的一类色谱柱。 在反相色谱中极性-离子型化合物是在死体积附近被洗脱的,即不在反相柱上保留,所以对此类物质的分析是很困难的。在反相色谱上开发方法时,很重要的需求就是物质要在固定相上有保留,并且防止/减少硅胶表面硅羟基对化合物的吸附。为了使极性-离子型化合物能在反相柱上保留,我们经常会对化合物进行衍生(2)或使用离子对试剂(3)。在前一种方法中需要将极性-离子型化合物化学衍生为疏水性物质,而在后一种方法中则在流动相中加入带相反电荷的物质,使极性-离子型化合物变为中性物质。这两种技术不但比较繁琐和费时,特别是离子对技术会导致反相色谱不能与质谱(MS)及光散射检测联用,对实验造成很大限制。最近发展出来的硅胶制造技术可以使得固定相更适合极性化合物的保留(1)。硅羟基能在一定流动相条件下对极性化合物产生保留。另外一种方法就是对硅胶表面进行化学修饰,如图1 所示。http://ng1.17img.cn/bbsfiles/images/2011/04/201104121805_288426_1604317_3.jpg如果R 基团具有极性,如氰基(-CN),氨基(-NH2) 或二醇(-CH(OH)-CH2-OH),这样固定相就具有保留亲水化合物的能力。使用前一种典型的HILIC 柱对极性化合物的保留性质如图2a 所示。 在低有机相比例(高水相比例)时,亲水性化合物没有保留,因为亲水化合物更倾向于留在流动相中。当流动相的非极性最够强时(足够的有机相比例),极性化合物才会有保留。但是,典型的疏水化合物在HILIC 柱子上却没有保留。因此HILIC 柱可以分析一些极性化合物的混合样,但是当样品中既有极性化合物,又有非极性化合物时,极性化合物就会因没有保留而分不开。http://ng1.17img.cn/bbsfiles/images/2011/04/201104121806_288427_1604317_3.jpg一个用HILIC 柱子分离极性化合物的例子如图2c 所示(4)。显然这两种极性化合物在普通C18 柱上是没有保留的(Figure 2b)。如果分析对象只有极性化合物HILIC 柱子是很合适的选择,就像我们在图2b 和2c 中看到的一样。http://ng1.17img.cn/bbsfiles/images/2011/04/201104121807_288428_1604317_3.jpgANP,反反相: 尽管HILIC 柱通常能解决极性-离子型化合物的保留问题,但是却不能满足样品中同时有亲水和疏水物质存在的分析要求。事实上,反相柱也一样不能解决亲水、疏水同时存在的情况。但是,反反相(ANP)的柱子就可以解决亲水、疏水物质同时分离的难题,使色谱工作者不至于在反相与HILIC 柱之间做二选一的选择。“水相正相色谱,反反相”反映了这种柱子具有两种分离机制,这个名字说明反反相(ANP)具有流动相中含水的性质(反相分离机理),同时也具有正相色谱的保留机理(在流动相极性更弱情况下保留增加)。HILIC 柱只提供类似于正相的效果,单没有反相色谱的功能。事实上,ANP 的保留机理与反相与HILIC 有很大差异,接下来的章节就ANP 的分离效果进行论述。ANP 1: 图3a 展示了两种物质在ANP 柱上的保留图(一个在反相上有保留,一个在正相上有保留)。在此例中,两种保留机理显示的很清楚,并且区域是两种化合物都有保留的。在这种情况下,只要改变流动相就可以在反相色谱与正相色谱间进行转换。流动相中水的比例高,疏水物质被保留,而亲水物质不保留;流动相中有机溶剂比例高,亲水物质被保留,而疏水物质不保留。http://ng1.17img.cn/bbsfiles/images/2011/04/201104121808_288429_1604317_3.jpg图3b 显示了3 种化合物在高水相比例中的分离(反相机理)。在这个例子里,ANP 柱的分离行为就如同一根普通的反相柱。ANP 2: 图4a 显示了ANP 柱从两种物质得到的保留图的另外一种性质,在这种情况下,流动相的组成使得两类化合物都有很强的保留能力,这样极性和非极性化合物就可以同时在ANP 柱上实现分离。另外一种操作就是我们可以在分析亲水和疏水化合物时使用梯度洗脱的方法。与HILIC 柱相比(只有极性化合物在高有机相情况下保留)或者ANP1 情况(化合物的保留取决于有机相比例)ANP2 则提供了独特的分离能力,这种分离能力在商品化柱子中是很少见的。图4b 显示了在ANP2 洗脱模式下分离混合物的例子,两个化合物,一个是极性的(甲福明二甲双胍,Metformin),另外一个是非极性的(格列本脲,Glyburide)在Si-H 基础上的C18柱上的分离。在上图中流动相为50:50(乙腈:水),反相机理起主要作用,格列本脲的保留比极性化合物甲福明二甲双胍更强。中间的图是流动相比例为80:20(乙腈:水),正相机理强于反正机理,甲福明二甲双胍在格列本脲之后被洗脱(使用LCMS 确认)。当乙腈比例继续提高到85%,正相机理就会起主要作用,强极性的甲福明二甲双胍保留时间会比格列本脲长更多。http://ng1.17img.cn/bbsfiles/images/2011/04/201104121810_288430_1604317_3.jpgANP 3: 第三种分离模式如图5 所示,分析物为两性物质,这类物质多为大分子物质,而且同时含有一个/多个疏水及亲水基团,例如一些多肽或蛋白。在这种情况下,色谱工作者可以根据混合物中分析物的性质选择使用反相还是ANP 模式来进行分离。这一非同一般的能力为实验条件提供了很大的改变空间。图5b 提供的是一个化合物在不同流动相组成比例条件下按反相和正相机理进行保留的结果。这一系列色谱图的结果和图5a 中所预示的结果是一致的。与预期一致的是,当乙腈比例增加时,保留时间由一个最小值,这个最小值就是保留机理从反相变为ANP 的临近点。http://ng1.17img.cn/bbsfiles/images/2011/04/201104121811_288431_1604317_3.jpg具有ANP 保留性质的色谱柱: 最近,已经有使用Si-H 表面的固定相色谱柱开始使用(MicroSolv 公司的TYPE-C Silica 柱),这种色谱柱显示出具有我们之前所论述的ANP 性质和分离能力,如图3b,4b 和5b 所示,也同时被文献(5,6)所报道。TYPE-C Silica 色谱柱固定相表面的组成与普通色谱柱的差异如图6 所示(Si-H 键的覆盖率为95%)。目前,与HILIC 柱一样,反反相的机理还不是完全清楚。TYPE-C Silica 这种Si-H 型固定相还具有其他优异性质:可以不需要从流动相中完全去除水就可以进行正







 我要推广仪器
我要推广仪器
 下载APP
下载APP