前两天有位外单位的人问分光光度计测量含量时用面积还是峰值来定量,因为没有见过它的仪器,不知道具体是什么情况,不过个人认为还是峰值来定量好,表示是最大吸收。面积所有波长的吸收,还有有可能会有干扰。这样理解对吗?还是真有用面积还定量的仪器?
蜂蜜中碳4植物糖含量测定~稳定碳同位素比值质谱法,有人参加上述实验室比对吗?
怎么结合谱线的峰值看EDX的测试结果?如果测得的Pb含量很高,而Pb对应的峰很小,这怎么解释?
求教各位版友~对这台机器还是比较菜,求大家帮忙~~着急~~GCMS 6890-5973,做测量审核样品——大米粉中的毒死蜱含量测定使用内标法定量,在制作标准曲线时,各点的内标含量均为0.5ppm(毒死蜱标准品标明用丙酮稀释,内标为之前储存也,接手前就一直使用丙酮稀释)样品按照国标处理,加内标亦为0.5ppm,最终用正己烷定容1mL标准曲线点跑出的谱图峰值很低,内标峰值为500-,而样品所得谱图内标峰值为1100+怕是基质问题,做过如下标准品①将购买的毒死蜱标准品用丙酮稀释,加内标,用正己烷定容1mL②将购买的毒死蜱标准品用丙酮稀释,加内标,用丙酮定容1mL结果相同~由于大米粉中毒死蜱含量未知所以只能确定内标的峰值过低,现在谱图不方便放上来,晚些时候来放图,期限又要到了,着急T.T求大家帮忙~~谢谢!!~万分感激!!~========================================================================2013.11.13 关于此次出现问题的最终解决方法反馈:2013.11.08检测结果已经寄送出去,11.11被对方签收,目前结果还没有出来,出后会补发的~现只总结下该问题的最终解决办法~~虽然具体原因不清楚,不过希望写出来能够有点帮助~~测过几次都出现同样问题后,另一位同事根据她测苏丹红的经验,猜测是不是应该把配置好的标准品也过下柱子于是将标准品配置好后过柱子备用,同时防止再出现问题,更换了新的衬管标准品与样品上机后,两者内标峰值基本保持一个水平了~约为30,000+标准曲线R值较好,回收率试验三次,平均回收率为104%以上就是基本的情况哦~~换衬管后也有侧过没过柱子的标准品,与换衬管前相似所以,为什么过了柱子后峰值变大,原因不明不知大家有何高见~~
峰值不对原因

[img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903071706_137216_1631661_3.jpg[/img]小弟接触红外时间不久,请教大家一个问题QQ:57536786E-mail:sunzhimems@163.com愿听赐教!图是我的薄膜样品,分别从薄膜的二个面入射而做出来的红外图。简单看来他们的峰数基本一致,但是在指纹区他们的峰值相差了大约一半啊?教科书上说:峰值与跃迁几率的大小和振动偶极距变化的大小有关。跃迁几率大和振动偶极距变化大则吸收峰强。那么对与我做的同一种材料的二个侧面,他们的峰值相差一半,应该怎么解释一下那?! 还是由于跃迁几率,偶极距变化吗? 二个侧面的成分含量不一样吗?!
USP30中色谱法测含量(甘露醇)公式 :50C(ru/rs)C─标准溶液中USP甘露醇参比标准品的浓度,单位以mg/ml计。ru─样品溶液的色谱峰值。rs─标准溶液的色谱峰值。请问50是怎么来的?公式的意义是什么?
气相色谱-质谱法检测鱼肉中脂肪酸含量摘要:本实验通过气相色谱-质谱联用技术检测鱼肉中脂肪酸的组分并通过面积归一化法对各组分进行百分含量测定。 关键词:梭鲈鱼;气相色谱-质谱联用;脂肪酸 实验部分1、仪器:气相色谱—串联质谱仪(Agilent 安捷伦,7890B-7000B);2、试剂与标准品:正己烷、甲醇(Fisher Scientific 飞世尔);三氯甲烷、氢氧化钾(天津光复试剂厂)。3、样品前处理:称取10g左右的鱼肉置于索式提取器中,加入40mL氯仿回流4h,取出后旋转蒸发浓缩近干,加入5mL乙醚-正己烷(1:2)溶液,溶解脂肪后倒入25mL试管中,继续加入5mL氢氧化钾-甲醇溶液(甲酯化),振摇后加入5mL正己烷静置10min后,吸取上层正己烷过滤膜后放入自动进样瓶中待测。4、色谱与质谱条件:色谱柱:HP-5MS(30 m×0.25 mm×0.5 μm);载气:氦气(99.999%);恒流模式流速:1.0 mL/min;进样:1.0 μL,不分流;进样口温度:250 ℃;程序升温:80 ℃保持1min,以10 ℃/min升温至180 ℃,以5℃/min升温至
[font=-apple-system, system-ui, &][size=15px][color=#121212]如果没有的话,我怎么看产物是否合成,是用chemdraw算出来的分子质量来找测出来的质谱txt里有没有相对应的峰吗。目标产物M1的分子质量用chemdraw算出来是554.0175,但测出来的质谱只有551的峰,质谱上553,554的峰特别小,是说明产物量特别低吗[/color][/size][/font][font=-apple-system, system-ui, &][size=15px][color=#121212][img]https://pic2.zhimg.com/v2-73243f58e2f5e23be021b4edce8cd995_b.jpeg[/img][/color][/size][/font][font=-apple-system, system-ui, &][size=15px][color=#121212][img]https://pic2.zhimg.com/v2-80fc8fdd1c10a95f65189016357c2ce5_b.jpeg[/img][/color][/size][/font]
GB/T 18932.1-2002 蜂蜜中碳-4植物糖含量测定方法 稳定碳同位素比率法中用到的同位素质谱仪可以用普通[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质代替吗?同位素质谱仪大约多少钱?
液相色谱-串联质谱法检测食品中维生素D含量 如何用液相色谱-串联质谱法(LC-MS/MS)检测食品中的维生素D含量。这项技术可是现代食品分析中的佼佼者,能够帮助我们精准地掌握食品中的营养信息。 样品前处理 提取:首先,我们需要从食品样品中提取维生素D。这一步很关键,因为提取效率直接影响最终的检测结果。通常,我们会使用有机溶剂,比如乙腈或甲醇,来提取维生素D。 净化:提取后的样品往往含有很多杂质,这些杂质会影响检测结果。因此,我们需要对提取液进行净化处理。常用的净化方法有固相萃取(SPE)和液液萃取(LLE)。 浓缩:净化后的样品溶液需要进行浓缩,以提高维生素D的浓度。常用的浓缩方法有氮气吹干和旋转蒸发。 仪器操作 流动相选择:选择合适的流动相对分离效果至关重要。通常,我们会使用水和有机溶剂(如甲醇或乙腈)的混合物作为流动相,并根据需要添加少量酸或缓冲液。 色谱柱选择:选择适合的色谱柱也很关键。C18反相色谱柱是常用的选择,因为它对维生素D有很好的保留效果。 质谱条件:设置合适的质谱条件,包括离子源温度、喷雾电压、碰撞能量等。这些参数的优化可以大大提高检测灵敏度和特异性。 故障排除 峰形不好:如果发现峰形不好,可能是由于流动相比例不合适或色谱柱污染。尝试调整流动相比例或清洗色谱柱。 灵敏度低:如果灵敏度不够,可能是由于样品提取效率低或仪器参数设置不当。检查提取方法并优化仪器参数。 杂峰干扰:如果出现杂峰干扰,可能是由于样品净化不彻底或流动相选择不当。尝试改进净化方法或更换流动相。 仪器故障:遇到仪器故障时,首先要保持冷静,然后根据仪器的报错信息查找原因。必要时,可以联系仪器厂家进行维修。 总之,液相色谱-串联质谱法检测食品中维生素D含量是一项复杂但非常重要的技术。通过掌握这些操作要点和故障排除方法,我们可以更加准确、高效地完成检测任务。
请教各位,全血中EDTA含量检测有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的检测方法吗,如果配制标准曲线,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]连用法检测全血中EDTA含量,可以直接进质谱吗。谢谢
[table=100%][tr][td]在做样品光谱分析时,发现含有相同官能团的样品吸收峰峰值呈现出左移变大的规律,原因怎么分析呢?[/td][/tr][/table]
大家好: 现在我碰到一个问题,在用GC1690色谱,用FID检测产品时,发现主峰前的杂质峰含量明显偏低,如果要使其杂质含量高,应怎么调色谱? 谢谢!
1 试剂和对照品除特殊说明,所用试剂均为色谱纯。1.1 试剂:甲酸,甲醇,氯化钠(分析纯),乙腈1.2 对照品:邻苯二甲酸酯类混合对照(供含量测定用)、DNP(供含量测定用)2 仪器2.1 液相质谱联用仪(API3200)2.2 分析天平2.3 超声震荡器2.4 涡旋仪2.5 离心机2.6 氮吹仪3 分析方法3.1 色谱参考条件1-适用于DNP3.1.1 色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长5cm,内径4.6mm,粒径2.7μm )或同等性能的色谱柱3.1.2 柱温:40℃3.2 色谱参考条件2-适用于剩余的22种塑化剂3.2.1 色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长5cm,内径4.6mm,粒径2.7μm )或同等性能的色谱柱与以十八烷基硅烷健合硅胶为填充剂(柱长15cm,内径4.6mm,粒径5μm )或同等性能的色谱柱两根串联3.2.2 柱温:40℃3.2.3 流动相:时间(分钟)流速(ml/min)0.1%甲酸水溶液(%)甲醇(%)0.000.820802.000.820805.000.820808.000.829813.000.829813.101.229814.001.229814.011.329818.001.329818.100.8208023.000.820803.2.4 质谱条件:ESI+ 模式化合物 Q1Q3Time(msec)IDDPEPCECXP邻苯二甲酸酯二壬酯419.335149.100300DNP 166.06.0021.004.00 419.335275.200300DNP 260.45.6215.356.25邻苯二甲酸酯二甲酯195.116320DMP 11510163 195.113520DMP 21510353邻苯二甲酸二乙酯223.1149.120DEP 12010273 223.1121.120DEP 22010443邻苯二甲酸二丙酯251.214920DPrP12310173 251.2191.120DPrP22310173邻苯二甲酸二异丙酯251.214920DIPrP12310173 251.2121.120DIPrP22310173邻苯二甲酸二烯丙酯247.1189.120DAP 12010123 247.1149.120DAP 22010263邻苯二甲酸二异丁酯279.214920DIBP 12310223 279.2205.120DIBP 22310123邻苯二甲酸二丁酯279.214920DBP 12310223 279.2205.120DBP 22310123邻苯二甲酸二(2-甲氧基)乙酯283.2207.120DMEP 12810113 283.259.120DMEP 22810303邻苯二甲酸二异戊酯307.27120DIPP13010253 307.2149.120DIPP23010213邻苯二甲酸二(4-甲基-2-戊基)酯335.314920BMPP 13010273 335.3167.120BMPP 23010183邻苯二甲酸二(2-乙氧基)乙酯311.2221.120DEEP 13010123 311.273.120DEEP 23010213邻苯二甲酸二戊酸307.2149.120DPP13010213 307.2219.120DPP23010123邻苯二甲酸丁基苄基酯313.291.120BBP 13010403 313.2149.120BBP 23010213邻苯二甲酸二己酯335.314920DHXP 13010273 335.3233.120DHXP 23010123邻苯二甲酸二(2-丁氧基)酯367.3101.120DBEP 13510183 367.3249.120DBEP 23510123邻苯二甲酸二环己酯331.2149.120DCHP 13010363 331.2167.120DCHP 23010213邻苯二甲酸二庚酯363.214920DHP 13510223 363.2247.120DHP 23510133邻苯二甲酸二(2-乙基)己酯391.314920DEHP 13510253 391.3167.120DEHP 23510203邻苯二甲酸二苯酯319.122520D_P 13010163 319.17720D_P 23010543邻苯二甲酸二正辛酯391.314920DNOP 13510253 391.3261.220DNOP 23510133邻苯二甲酸二异壬酯419.314920DINP 13810543 419.3127.220DINP 23810183邻苯二甲酸二异癸酯447.214120DIDP 14510153 447.2289.220DIDP 24510153CURCADISTEMGS1GS2256550065065653.3 溶液制备3.3.1对照品溶液制备:3.3.1.1邻苯二甲酸酯二壬酯(DNP)对照液:精密称取邻苯二甲酸酯二壬酯(DNP)对照品5mg,置于500ml容量瓶中,加入适量甲醇超声溶解,定容。精密量取1ml置于100ml容量瓶中,加入甲醇定容至刻度,摇匀,过0.45μm的滤膜,即得。3.3.1.2邻苯二甲酸酯类混合对照液:精密量取邻苯二甲酸酯类混合对照品1ml,置于100ml容量瓶中,加入甲醇定容至刻度,摇匀,过0.45μm的滤膜,即得。3.3.2供试品溶液制备:3.3.2.1不含油脂固体或半固体试样 取固体试样20粒(片)或10g(若试样为胶囊,应取内容物),研细,混合均匀。精密称取试样1.0g,置于50ml具塞比色管中,精密量取5ml乙腈,超声(功率500W,频率50kHz)处理至提取完全(约5min),取上清液,经0.45μm的滤膜过滤,即得。3.3.2.2 含油脂试样 精密称取混合均匀试样1.0g,置于25ml具塞磨口玻璃试管中,加入5ml乙腈,涡旋混合2min, 超声(功率500W,频率50kHz)处理至提取完全(约10min),离心,收集上清液。重复提取一次,合并上清液。将提取液于40℃氮吹至干,精密量取1ml乙腈,超声(功率500W,频率50kHz)处理至提取完全(约1min),放入-18℃的冰箱冷藏2h,离心,取上清液,经0.45μm的滤膜过滤,即得。3.4 测定精密吸取对照品溶液进样量:2μL,4μL,6μL,8μL,10μL与供试品溶液进样量:10μl,注入液相质谱联用仪,建立标准曲线方程。供试品色谱中应呈现与对照品色谱峰保留时间相同的色谱峰定量。3.5 结果计算X=V×C×100/M式中: X—试样中邻苯二甲酸酯类的含量,mg/100g;C—试样溶液中邻苯二甲酸酯类的浓度,mg/mL; M—试样的质量,g; V—试样稀释的体积,mL。
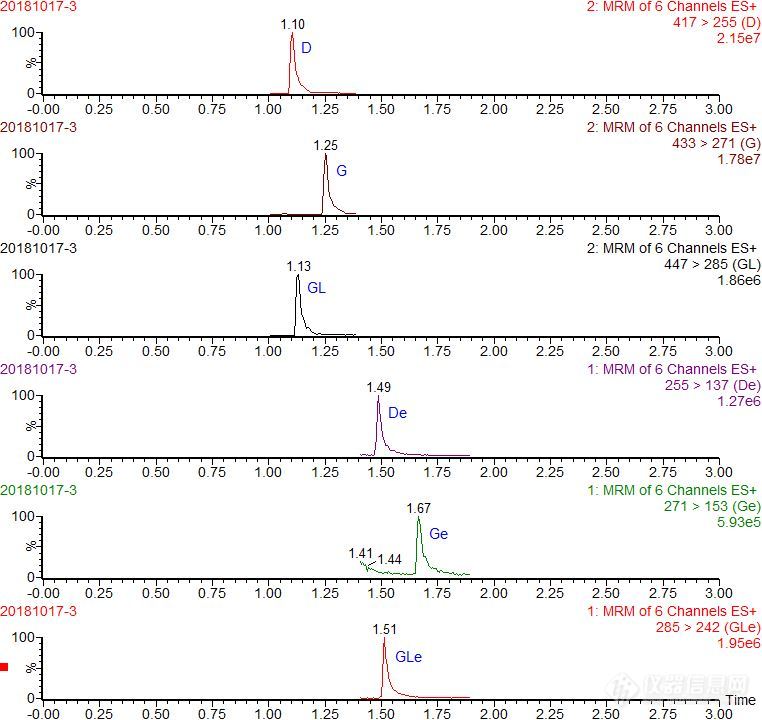
[align=center][b]超高效液相色谱-串联质谱法测定大豆中大豆异黄酮的含量[/b][/align][align=center]户江涛[/align][align=center](农业农村部豆类产品质量安全风险评估实验室(佳木斯),黑龙江省农垦科学院测试化验中心,黑龙江佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了检测大豆中大豆异黄酮含量的分析方法。试样经90%甲醇水提取后,6种大豆异黄酮在C[sub]18[/sub]色谱柱上以0.1%甲酸水溶液和乙腈为流动相,进行液相色谱分离;质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,6种大豆异黄酮分别在0.01~0.5 mg/L(D、GL、G)和0.002~0.1 mg/L(De、GLe、Ge)范围内线性关系良好,相关系数(R)为0.9993~0.9998,定量限(LOQ)为0.0001 g/kg。在大豆空白样品添加浓度分别为0.01、0.05、0.2 g/kg(De、GLe、Ge)和0.2、1、2 g/kg(D、GL、G),6种大豆异黄酮的平均回收率为86.6%~96.2%,相对标准偏差(RSD)为1.07%~5.93%(n=6)。本方法简便、灵敏、抗干扰,适用于大豆中大豆异黄酮含量检测。关键词:超高效液相色谱-串联质谱;大豆;大豆异黄酮[align=center]Determination of soybeanisoflavone in soybean by ultra performance liquid chromatography-tandem massspectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Laboratory of Qualityand Safety Risk Assessment for Soybean products, Ministry of Agriculture andRural Affairs, Testing and Analysis Center of Heilongjiang Academy of LandReclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A methodwasdeveloped for the determination of soybeanisoflavone in soybean by ultra performance liquid chromatography-tandem massspectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). The samples were extracted by 90% methanol-water, then 6 soybean isoflavones were separated on aWaters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of0.1% formic acid and acetonitrile, and finally detected by positive eletrosprayionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reactionmonitoring(MRM) mode. The results showed the linearities of 6 soybean isoflavones were good in the concentrationrange of 0.01~0.5 mg/L(D、GL、G)and 0.002~0.1 mg/L(De、GLe、Ge), the correlation coefficients were 0.9993~0.9998. The limitof quantification(LOQ) of soybean isoflavone was 0.0001 g/kg. At the spiked levels of 0.01、0.05、0.2 g/kg(De、GLe、Ge)and 0.2、1、2 g/kg(D、GL、G) in the blank soybean samples, the mean recovery of soybeanisoflavone was 86.6%~96.2%, andthe relative standard deviation(RSD) was 1.07%~5.93%(n=6).This method is simple,sensitive, anti-jamming and suitable for simultaneous determination of soybean isoflavone in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem massspectrometry (UPLC-MS/MS) soybean soybean isoflavone大豆异黄酮(soybean isoflavone)是一族化合物的统称,是大豆植物体内的一种次生代谢产物,是大豆主要活性成分之一,其母核为3-苯并吡喃酮,主要包括大豆苷、大豆黄苷、染料木苷及其相应苷元[sup][/sup]。研究表明,大豆异黄酮除具有天然抗氧化作用外[sup][/sup],还具有降低胆固醇含量、预防多种癌症及改善妇女更年期综合征等多方面生物功效[sup][/sup]。大豆异黄酮主要存在于大豆籽实中,其总含量约为0.4~5 g/kg,其中大豆苷、大豆黄苷和染料木苷这三种含量约占总量的97%~98%,而其对应的苷元含量仅占2%~3%左右[sup][/sup]。目前,大豆异黄酮的检测方法主要有高效液相色谱法(HPLC)[sup][/sup]、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]、紫外分光光度法[sup] [/sup]、质谱法(HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS[sup][/sup])等。紫外分光光度法[sup] [/sup]只能测定大豆异黄酮总量,且灵敏度不高;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]需要对异黄酮进行衍生,前处理复杂;目前,大豆异黄酮检测现有的国家标准GB/T 26625-2011[sup] [/sup]采用的是高效液相色谱法(HPLC),在实际检测过程中发现,由于紫外检测器灵敏度不高,存在个别样品中异黄酮相应苷元检测不到的情况;同时大豆提取液中含有蛋白、脂肪等杂质影响色谱柱柱效,以至于不能满足分离度要求,严重干扰低含量组分峰面积积分定量。而[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法质谱检测器灵敏度高,通过选定大豆异黄酮的特征离子,能有效去除上述杂质干扰,定量更加准确可靠。目前,国内外采用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法检测大豆中大豆异黄酮含量的文献很少[sup][/sup]。本文对大豆中大豆异黄酮检测的前处理方法借鉴GB/T 26625-2011[sup][/sup],提取液改用UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS测定。该方法前处理过程简便、灵敏度高、分析时间短、抗干扰能力强,适用于大批量大豆样品中大豆异黄酮含量的检测。[b]1 实验部分[/b]1.1 材料与试剂大豆苷(daidzin,记为D,以下同)、大豆黄苷(glycitin,GL)、染料木苷(genistin, G)、大豆素(daidzein,De)、大豆黄素(glycitein, GLe)、染料木素(genistein,Ge)(纯度≥99%,Dr.Ehrenstorfer公司);甲醇、乙腈、甲酸(色谱纯,Fisher公司);实验用水为Millipore纯水仪制备。1.2 仪器与设备Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);KQ-500DE型超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司);CR21GⅢ型高速离心机(HITACHI公司)。1.3 大豆异黄酮标准储备液的配置分别称取适量的D、GL、G、De、GLe、Ge标准品,用甲醇配置成质量浓度为1mg/mL标准储备液,于-18℃冰箱保存(有效期6个月),待用;使用时用10%甲醇水逐级稀释成所需浓度的混合标准工作液,现用现配。1.4 样品前处理提取:称取粉碎均与后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0 mL90%甲醇水,涡旋混合30 s后置于60℃超声波清洗器中提取30 min,在离心机中以15000 r/min离心5 min,将上清液转移至100 mL容量瓶中,残渣再加入10.0 mL90%甲醇水溶液按上述步骤提取后,合并两次上清液于100 mL容量瓶中,用10%甲醇水溶液定容至刻度,摇匀。a)De、GLe、Ge的测定:取1 mL过0.22um有机系微孔滤膜,供UPLC/MS/MS分析测定;b)D、GL、G的测定:由于D、GL、G含量较高,需要将a)中过完滤膜的待测液用10%甲醇水稀释50倍后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:1μL;流动相A:乙腈;流动相B:0.1%的甲酸水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1.1min,100% A~10% A,4.1 ~5.0min 10% A。质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1大豆异黄酮的质谱参数[/align][align=center]Table 1 MRM parameters of soybean isoflavone[/align] [table][tr][td] [align=center]Analyte[/align] [/td][td] [align=center]Cone/V[/align] [/td][td] [align=center]Parent ion/(m/z)[/align] [/td][td] [align=center]Daughter ion/(m/z)[/align] [/td][td] [align=center]Collision energy/V[/align] [/td][/tr][tr][td] [align=center]D[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]417[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]255﹡[/align] 137[/td][td] [align=center]27[/align] [align=center]18[/align] [/td][/tr][tr][td] [align=center]G[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]433[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]271﹡[/align] 153[/td][td] [align=center]21[/align] [align=center]50[/align] [/td][/tr][tr][td] [align=center]GL[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]447[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]285﹡[/align] 270[/td][td] [align=center]25[/align] [align=center]46[/align] [/td][/tr][tr][td] [align=center]De[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]255[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]137﹡[/align] 181[/td][td] [align=center]30[/align] [align=center]26[/align] [/td][/tr][tr][td] [align=center]Ge[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]271[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]153﹡[/align] 215[/td][td] [align=center]30[/align] [align=center]25[/align] [/td][/tr][tr][td] [align=center]GLe[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]285[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]242﹡[/align] 168[/td][td] [align=center]27[/align] [align=center]35[/align] [/td][/tr][/table]﹡quantitativeion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(纯水、乙酸铵溶液)分别与甲醇、乙腈的流动相体系组合,结果发现目标物在酸性体系中比非酸性体系响应更高、峰形更好;同时大豆提取液中含有蛋白、脂肪等杂质可能会残留在色谱柱上,影响色谱柱的使用寿命,而乙腈比甲醇体系洗脱能力更强,可以有效去这些杂质。综合考虑目标物信号强度、色谱分离效果以及除杂等因素,本研究采用0.1%甲酸水溶液+乙腈流动相体系。质谱的选择:根据6种大豆异黄酮的分子量,用10%甲醇水配置1.0 mg/L 大豆异黄酮标准溶液直接注射到质谱中,在正离子模式下分别对各种组分进行母离子及对应子离子全扫描,最终确定的质谱条件见表1。2.2 质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)与色谱法(HPLC)的比较国家标准《GB/T 26625-2011粮油检验大豆异黄酮含量测定高效液相色谱法》[sup][/sup]中规定的大豆异黄酮检测方法为HPLC法。对同一大豆样品分别采用本文UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法(MRM色谱图见图1、2)和GB/T 26625 HPLC法检测,结果表明这两种方法测定的大豆异黄酮总含量值基本一致。由于De、GLe、Ge这三种苷元在大豆中含量很低,用HPLC法检测时,紫外检测器灵敏度不高,存在个别样品中上述三种组分检测缺失的情况;同时在实际大批量样品检测中发现,随着进样次数的增加,色谱柱柱效下降,大豆提取液中存在的蛋白、脂肪等杂质对含量低的目标物峰干扰越来越大,定量困难。研究发现,同浓度的大豆异黄酮在质谱检测器上的响应值要远远超过紫外检测器,同时质谱法可以通过选定大豆异黄酮的特征离子,有效地去除杂质的干扰,其目标物分离度不受色谱柱进样次数增加的影响,定量更加准确可靠。[align=center][img=,690,651]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050912587968_4111_3299836_3.jpg!w690x651.jpg[/img][/align][align=center]图1 大豆异黄酮标准溶液(0.01mg/L)MRM色谱图[/align][align=center]Fig.1 MRM chromatograms of soybean isoflavone standard solution at 0.01 mg/L[/align][align=center][/align][align=center][img=,690,653]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050913342201_5843_3299836_3.jpg!w690x653.jpg[/img][/align][align=center]图2 大豆样品中大豆异黄酮MRM色谱图[/align][align=center]Fig.2 MRM chromatograms of soybean isoflavone in soybean[/align]2.3线性范围和定量限吸取不同体积的大豆异黄酮标准储备液(1.3),用10%甲醇水分别配置0.002、0.005、0.01、0.05、0.1(De、GLe、Ge)和0.01、0.05、0.1、0.2、0.5(D、GL、G)的大豆异黄酮上机混合标准溶液,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,大豆异黄酮标准溶液在各自浓度范围内线性良好,相关系数R为0.9993~0.9999。以10倍信噪比(S/N)计算,大豆异黄酮上机液最低定量浓度为0.001 mg/L,通过公式(1)计算得到大豆中大豆异黄酮含量,最终确定本方法大豆异黄酮的定量限(LOQ)为0.0001 g/kg。糠氨酸质量分数计算公式:[align=center][img=,207,87]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050915166414_5621_3299836_3.jpg!w207x87.jpg[/img] ………………(1)[/align] 式中:X为试样中大豆异黄酮含量,以g/kg计;C为大豆异黄酮上机浓度(mg/L);V为定容体积(V=100)。表2 大豆异黄酮标准溶液的线性方程和相关系数[align=center]Table 2 Linear equation and correlation of soybean isoflavone in 10% methanol-water standard solutions[/align] [table][tr][td] [align=center]Analyte[/align] [/td][td] [align=center]Linear range/(mg/L)[/align] [/td][td] [align=center]Linear equation[/align] [/td][td] [align=center]R[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]D[/align] [align=center]GL[/align] [align=center]G[/align] [align=center]De[/align] [align=center]GLe[/align] [align=center]Ge[/align] [/td][td] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [/td][td] [align=center]Y=2393.6x+479.38[/align] Y=1885x+139.66 [align=center]Y=1470.9x+187.97[/align] [align=center]Y=4287.9x+442.79[/align] [align=center]Y=3521.7x-103.62[/align] [align=center]Y=1993x+122.79[/align] [/td][td] [align=center]0.9995[/align] [align=center]0.9999[/align] [align=center]0.9993[/align] [align=center]0.9998[/align] [align=center]0.9997[/align] [align=center]0.9998[/align] [/td][td] [/td][/tr][/table]2.4回收率和精密度大豆中De、GLe、Ge含量较低,而D、GL、G含量较高,故本方法准确度实验分为高低浓度梯度组进行加标。称取大豆试样1.00 g,分别添加0.01、0.05、0.2 g/kg(De、GLe、Ge)和0.2、1、2 g/kg(D、GL、G),每个水平重复6次,同时做该大豆的空白本底实验。按照1.4前处理方法处理后上机检测,计算回收率(扣除空白),结果表明:不同添加浓度下,De、GLe、Ge的平均回收率为91.7%~96.2%,相对标准偏差(RSD,n=6)为2.78%~5.93%;D、GL、G的平均回收率为86.6%~93.8%,相对标准偏差(RSD,n=6)为1.07%~3.77%。[b]3 结语[/b]本文建立了超高效液相色谱-串联质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)测定大豆中大豆异黄酮含量的分析方法。该方法灵敏度高,线性范围宽,能同时覆盖大豆中多梯度浓度大豆异黄酮组分含量的测定。同时该方法具有较高的准确度和精密度,前处理步骤简单,分析速度快,可有效避免由于色谱柱柱效下降对最终检测结果的影响,特别适合大批量样品的检测。田娟娟, 宋宏哲, 张飞, 等. 水剂法纯化大豆异黄酮的研究. 大豆通报, 2005, 6:19-22. Hagen M K, Ludke A, Araujo A S, et al.Antioxidant characterization of soy derived products in vitro and the effect ofa soy diet on peripheral markers of oxidative stress in a heart disease model .Canadian Journal of Physiology and Pharmacology, 2012,90(8):1095-1103. 徐春华, 张治广, 谢明杰, 等. 大豆异黄酮的抗氧化和抗肿瘤活性研究研究 . 大豆科学, 2010, 29(5): 870-873. 李俏俏, 王清路, 薛金艳, 等. 大豆异黄酮对绝经女性血清中脂类物质的影响的研究 . 大豆科学, 2009, 28(1):172-174. 胡润芳, 张玉梅, 陈宇华, 等. 大豆异黄酮含量的初步研究. 东南园艺, 2017, 6:9-11. 刘琴, 朱媛媛, 白兴梁. 不同种类大豆中大豆异黄酮含量及抗氧化性比较. 北京工商大学学报(自然科学版), 2012, 30(6): 45-51. 袁凤杰, 姜莹, 董德坤, 等. 中国大豆核心种质异黄酮含量分析.中国粮油学报, 2011, 26(2):5-8. Tepavcevic V, Atanackovic M,Miladinovic J,et al. Isoflavone composition,total polyphenolic content,and antioxidant activity in soybeans of different origin. MedFood,2010,13(3):657-664 GB/T 26625-2011《粮油检验大豆异黄酮含量测定高效液相色谱法》. Liggins J,Bluck J C. Deidzein and genistein content of fruits and nuts. Journal ofNutritional Biochemistry,2000,11(6):326-331. 鞠兴荣, 袁建, 汪海峰. 三波长紫外分光光度法测定大豆异黄酮含量的研究. 食品科学, 2001, 22(5):46-48.
请问,无机颗粒填充环氧树脂复合材料的DMA图谱中,损耗角正切值的高度代表阻尼行为,那么填料经过表面处理,参与环氧树脂交联体系,那么损耗角正切峰值高度是增高还是降低?我的理解是:体系交联度增大,刚性增强,阻尼减小,损耗角正切峰值高度应该降低;但是,另外一个角度考虑,界面作用力增大,内摩擦增大,阻尼行为应该增大啊(即损耗角正切峰值高度增高)请大家看看哪种理解是对的?
我在做银杏叶片萜类内酯含量测定时,流动相是正丙醇四氢呋喃和水,标准规定对照品溶液(甲醇溶剂)进5和20ul,进5ul峰型很正常,柱效很高,但是在进20ul的时候,峰就变形了,上坡像连绵起伏的山峰,下坡倒正常,后来我进了一针样品(丙酮溶剂,20ul),个个峰都变形。但是非常奇怪的是,我在不久前刚做了这项含量,各个条件基本是一模一样的,连对照都是同一份溶液,对照在进20ul时峰型很正常。柱子不可能有问题,而且换了一根也是同样问题,流动相是配在一起的。1、请教一下,我的流动相是配在一起的,会不会有可能吸滤头脏了。2、另外,我曾在别的液相和蒸发光检测器上按上述方法测过,峰型很好,会是什么原因。
:高效液相色谱_串联质谱法测定酸奶中纳他霉素的含量期刊:中国卫生检验杂志作者:李媛媛; 王伟; 关树文; 李刚; 董巧红;链接:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=ZWJZ201004027
分子量测定: Q:为什么测定的结果跟理论的结果不一致? A:因为机器测定会有一定的误差,如100000Da的蛋白质最大误差是100Da,如果测定的结果与理论值成倍数关系的可能原因是:0.3倍可能是三电荷峰,0.5倍可能是双电荷峰,原因是样品含碱氨基酸较多;2倍可能是二聚体,3倍可能是三聚体,原因可能是样品中含有过多的二硫键。 Q:为什么我的样品非常纯,但在主峰的附近会有一些小峰?同时在主峰一半或两倍的地方有一个小峰? A:这是因为在激光的扫描的过程中可能会使样品分子损失一些离子,如H2O、NH3等,或者出现样品分子与基质的加合峰,正常的情况下MALDI-TOF单电荷峰占主要,所以有时会在主峰一半或两倍的地方有一个小峰,是双电荷峰和二聚体峰; Q:为什么我的样品纯度、浓度都很好,盐浓度也很低,却得不到结果? A:这只能与蛋白质本身的结构有关,基质不能很好的分离样品,所以希望您能尽可能滇供样品其他的理化质,如酸、碱蛋白、糖蛋白等。的确,有时您的样品的所有质、状态都正常,还是不能得到您想要的结果,这种问题其实已经超出了我们能力的范围,我们仅仅是您整个实验的一个环节,能够做到的是向您提供一个准确的结果。 肽指纹图谱及数据库检索 Q:为什么考马斯染色的结果比银染的结果好。 A:主要是银染的灵敏度较高,得到的蛋白质量较少,再加上在处理样品的过程中银染要脱银,增加洗脱的次数,样品的损失较大,所以噪音和角蛋白污染对目的蛋白的影响较大,对最终的数据库检索也会产生影响 Q:我的PMF的峰值较少,为什么?这些片段数量及峰值是否达到最基本的数据库检索的要求?影响检索的结果吗? A:因为我们用的是胰酶对蛋白质进行降解的,胰酶是一种专一较强的酶,只水解精氨酸、赖氨酸的C端,可能是你的样品的酶解的位点偏多或偏少,偏多酶解为小片段,峰值跟基质在一起难以分辨,偏少肽段的分子量偏大,胶内的样品滞留在胶内,在质谱图上体现不出来,溶液样品可以收集到,但是分子量越大检索的结果越不准确;理论上图谱的峰越多越好,要是都能匹配上那么蛋白的确定程度就更高,相反就越低;一般至少要拿到4个肽段。 Q:如何理解这些片段峰值大小意义,越高(纵坐标值大)越精确吗?以及这些酶解片段的数量及峰值一般应达到什么标准才算比较好?? A:片段峰横坐标的大小的意义是酶解下来的肽段的质量大小,纵坐标表示的收集的该肽段的相对的信号的强度,左边是相对的,右边是绝对的,绝对的越高,抗干扰的能力越好,质谱的精确度由仪器来决定的。我们的仪器的精确度在10ppm以内。 Q:图谱上肽段多于8条,为什么选这8条? A:至于提交检索的肽段的数量是根据肽段的信号强度来选取的,同时在软件处理的过程中会过滤一部分峰, 如胰酶的自解峰等; Q:数据库检索报告中m/zsubmitted,MH+matched两项是什么意思? A:m/z submitted 的意思是您提交给数据库的m/z值(因为质谱仪用m/z来分离样品的), MH+matched指的是与你提交数值相匹配数值,因为在样品在离子化过程中从基质中得到一个质子,所以它会加一个H Q:这两项的分子量和你们的图谱上相应分子量不一致。 A:因为在用图谱数据在数据库检索之前需要通过一个数据的校正和处理,所以图谱上的值与提交检索的值有区别图谱中的峰值是经过一个软件处理后的结果,主要是经同位素峰的合并图谱上的结果会不一至的,例如在你的图上读到的是1519.402,在看图的时候把它给放大处理(只有我这里的图谱处理的软件才能看到),就会发现存在1518.402、1520.402、1521.402、1522.402等相差1的质谱峰,这是由于在天然界中好多物质都存在同位素如O16、O17、O18等,所以在提交给数据库之前会用一个类似于碳水化合物的通式进行校正,所以图谱上读到的数可能会与提交的相差1,还在处理的过程的设置也会影响给出的峰值,但应该在0.1Da以内的;同时在还会进行胰酶自解峰的扣除和空白对照峰的扣除。
T/BPCT 001-2023牛乳及制品中A2 β-酪蛋白含量的测定 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]质谱法
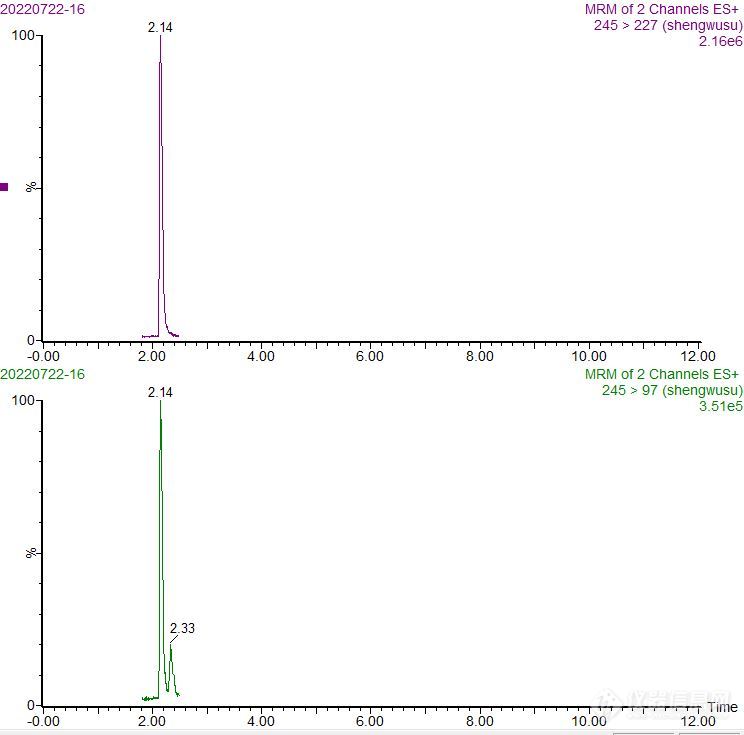
[size=16px]超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定奶粉中生物素的含量[/size][align=center][size=16px]户江涛[/size][/align][align=center][size=16px](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/size][/align][size=16px]摘要:采用超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法建立了检测奶粉中生物素含量的分析方法,对试样提取、净化条件,流动相、色谱柱和质谱条件进行了优化,结果表明该方法与国标微生物法对同一样品检测得到的生物素含量基本一致,但检测所需时间大大减少,且抗干扰能力、精密度均比微生物法高,特别适和大批量奶粉中生物素含量检测。关键词:超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱;奶粉;生物素生物素又称维生素B7,是生物体内羧基转化酶作用的一种辅酶,在人体生长、代谢、发育过程中发挥着重要的作用。人类自身不能合成生物素,需从膳食中获得,而奶粉是人类(特别是婴幼儿)获取生物素的重要途径,准确测定奶粉中生物素含量有重要意义。目前国家标准规定的生物素测定方法《GB 5009.259-2016 食品安全国家标准 食品中生物素的测定》为微生物法。该方法需要购买特定菌种,成本较高,且菌种难保存、易受污染,实验操作复杂、费时费力、技术难度大、对检验人员和实验室要求较高,且容易受到基质干扰、检测结果重复性较差。同时奶粉成分复杂,所含生物素含量极低,一般为十几个微克/100克。因此,制定一种准确、高效、便捷、灵敏度高的生物素测定方法迫在眉睫。基于高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]的分离能力和质谱的高灵敏度、高选择性,采用[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱测定法具有前处理简单、分析速度快,适用的基质范围广、实用性强,可以为奶粉中生物素含量的测定提供一种有效的检测手段。本文建立的超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定奶粉中生物素含量[color=black]的方法前处理过程简便、分析时间短、灵敏度高、抗干扰能力强,特别适用于大批量奶粉样品中生物素[/color]含量的检测。1 实验部分1.1 材料与试剂[color=black]生物素(纯度[/color][font=宋体][color=black]≥[/color][/font][color=black]99%,Sigma公司);婴儿配方乳粉定量分析质控样品(BQC1051147452,北京普天同创生物科技有限公司);乙腈、甲酸(色谱纯,Fisher公司);Prime HLB固相萃取柱(200 mg,3 mL,[/color][font=宋体]Waters[/font][color=black]公司);0.2 um有机系滤膜;实验用水为Millipore纯水仪制备。[/color]1.2 仪器与设备UPLC XEVO TQ-S超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱仪(Waters公司);涡旋振荡器。1.3 [color=black]生物素[/color]标准储备液的配置称取一定量生物素[color=black]标准品[/color],用50%乙醇-水溶液配置成质量浓度为100 ug/mL标准储备液,于2~4℃冰箱保存(有效期1个月),待用;临用前将溶液回温至室温,并吸取一定体积储备液用水逐级稀释成所需浓度的标准工作液。1.4 样品前处理准确称取1.00 g(精确到0.01 g)奶粉试样于50 mL离心管中,加入10.00 mL纯水涡旋混匀2 min,然后加入10.00 mL乙腈,涡旋混匀1 min,然后在离心机中以15000 r/min离心5 min,取出后吸取2 mL上清液置于[color=black]Prime HLB固相萃取柱中,使其自然流出弃去最初几滴,然后用玻璃试管接取流出液约1 mL涡旋混匀,[/color]过0.22[font=宋体]u[/font]m有机系微孔滤膜后供UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析测定。1.5 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]及质谱条件[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]:色谱柱:Waters HSS [font=times new roman]T3(1.8 μm,100mm×2.1mm);柱温:30℃[/font];流速:[font=times new roman]0.3 [/font]mL/min;进样量:[font=times new roman]2[/font] [font=times new roman]μL;流动相A:乙腈;流动相B:0.1%的甲酸水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~10% A,4.1 ~5.0min 10% A。[/font]质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[/size][align=center][size=16px]表1生物素的质谱参数[/size][/align][table][tr][td][align=center][size=16px]分析物[/size][/align][/td][td][align=center][size=16px]锥孔电压/V[/size][/align][/td][td][align=center][size=16px]母离子/(m/z)[/size][/align][/td][td][align=center][size=16px]子离子/(m/z) [/size][/align][/td][td][align=center][size=16px]碰撞能量/V[/size][/align][/td][/tr][tr][td][size=16px]生物素[/size][/td][td][align=center][size=16px]30[/size][/align][size=16px][/size][/td][td][align=center][size=16px]245[/size][/align][size=16px][/size][/td][td][align=center][size=16px]227﹡[/size][/align][align=center][size=16px]97[/size][/align][size=16px][/size][/td][td][align=center][size=16px]13[/size][/align][align=center][size=16px]25[/size][/align][size=16px][/size][/td][/tr][/table][size=16px]﹡为定量离子2 结果与讨论2.1 色谱质谱条件及前处理过程的优化流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与甲醇、乙腈的流动相体系组合,结果发现生物素在乙腈体系中响应值比甲醇更好一些,故本研究采用0.1%甲酸水溶液+甲醇流动相体系。色谱柱的选择:比较了[font=宋体]Waters [/font]BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm)和[font=宋体]Waters [/font]HSS T[sub]3[/sub](1.8 μm,100mm×2.1mm)两种不同填料的分析柱,实验时发现目标物在这两款色谱柱上响应值差不多,但目标物在BEH C[sub]18[/sub]上保留时间比HSS T[sub]3[/sub]要短,考虑到生物素本身属于水溶性维生素,极性较强,若出峰太早可能造成奶粉中一些极性强的基质随目标物一起共流出进而干扰目标物测定,因此本方法采用了HSS T[sub]3[/sub]色谱柱。质谱参数优化:将1.0 mg/L 生物素标准溶液直接注射到质谱中,在正离子模式下进行母离子全扫描,发现目标物各自对应的准分子离子峰[M+H][sup]+[/sup]具有很好的响应,然后在分别进行子离子全扫描,各得到两对丰度高、干扰小的子离子对进行MRM监测,最终确定的质谱条件见表1,相应的色谱质谱图见图1、图2。前处理过程优化:生物素属于水溶性维生素,用纯水作为提取试剂可以得到很好的提取效果。但实验过程中发现,用纯水将奶粉溶解后整个溶液呈乳白色,只通过离心方式很难去除其中大量的蛋白、脂肪等杂质,需要对提取液进行除蛋白操作。通过考察乙酸铅、三氯乙酸、乙腈等几种常用的沉淀蛋白方法,综合考虑在去除蛋白的同时要尽可能减少其它杂质的引入,因此本方法采用乙腈除蛋白的方式,比较了几种不同水/乙腈比例,最终选定水/乙腈(1:1体积比)达到最优的实验效果。对于脂肪的去除则选用了目前较流行的[color=black]Prime HLB固相萃取柱通过式方法,即提取液通过Prime HLB时脂肪等大分子保留在SPE小柱上,目标物不保留以达到去除脂肪等杂质的目的,[/color]综合以上因素本实验最终采用了1.4的前处理方法。[/size][align=center][size=16px][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210071506558084_124_1729077_3.jpg[/img][/size][/align][align=center][size=16px]图1 [color=black]生物素[/color]标准溶液(10 ng/mL)MRM色谱图[/size][/align][size=16px][/size][align=center][size=16px][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210071506561980_1283_1729077_3.jpg[/img][/size][/align][align=center][size=16px]图2 奶粉样品中[color=black]生物素[/color]MRM色谱图[/size][/align][size=16px][color=black]2.2 线性范围和定量限[/color][color=black]吸取不同体积的生物素标准储备液(1.3),用[/color]纯水[color=black]分别配置不同浓度的[/color]上机标准溶液,以各自定量离子的峰面积(或与内标峰面积比值)为Y对应质量浓度X([color=black]m[/color]g/L)做标准曲线,得到的线性方程和相关系数见表2;以10倍信噪比(S/N)计算得到生物素的定量下限,结果见表2。表2 生物素标准溶液的线性方程、相关系数和定量下限(LOQ)[/size][table][tr][td][align=center][size=16px]分析物[/size][/align][/td][td][align=center][size=16px]线性范围/(ng/mL)[/size][/align][/td][td][align=center][size=16px]线性方程[/size][/align][/td][td][align=center][size=16px]R[/size][/align][/td][td][align=center][size=16px]LOQ/(ug/100g)[/size][/align][/td][/tr][tr][td][align=center][size=16px]生物素[sub] [/sub][/size][/align][/td][td][align=center][size=16px]0.2~50[/size][/align][size=16px][/size][/td][td][align=center][size=16px]Y=3078.1X-106.32[/size][/align][size=16px][/size][/td][td][align=center][size=16px]0.9993[/size][/align][size=16px][/size][/td][td][align=center][size=16px]0.5[/size][/align][/td][/tr][/table][size=16px][color=black]2.3回收率和精密度[/color][color=black]生物素在奶粉中天然存在[/color],选取已知生物素含量的奶粉作为基质进行加标。具体添加水平为:[color=black]0.5,5,50[/color] ug/100g。[color=black]每个[/color]水平重复6次,[color=black]同时做该奶粉的本底实验。[/color]按照1.4前处理方法处理后上机检测,回收率计算结果(扣除空白后)见表3。结果表明,该方法生物素的平均回收率为87.2%~110%,相对标准偏差(RSD,n=6)为2.3%~5.2%,均满足实验要求。[/size][align=center][size=16px]表3 奶粉生物素的加标回收率和相对标准偏差(n=6)[/size][/align][table][tr][td][align=center][size=16px]分析物[/size][/align][/td][td][align=center][size=16px]添加水平(ug/100g)[/size][/align][/td][td][align=center][size=16px]回收率/%[/size][/align][/td][td][align=center][size=16px]相对标准偏差/%[/size][/align][/td][/tr][tr][td][align=center][size=16px]生物素[/size][/align][size=16px][sub] [/sub][/size][/td][td][align=center][size=16px]0.5[/size][/align][align=center][size=16px]5[/size][/align][align=center][size=16px]50[/size][/align][/td][td][align=center][size=16px]86.8[/size][/align][align=center][size=16px]93.2[/size][/align][align=center][size=16px]91.6[/size][/align][size=16px][/size][/td][td][align=center][size=16px]4.6[/size][/align][align=center][size=16px]3.3[/size][/align][align=center][size=16px]2.1[/size][/align][size=16px][/size][/td][/tr][/table][size=16px][color=black]2.4实际样品分析[/color][color=black]为进一步验证该方法的准确性,采用本方法和《[/color]GB 5009.259-2016[color=black]》微生物法同时对北京普天同创生物科技有限公司的奶粉质控样品BQC1051147452生物素含量进行检测,结果见表4[/color]。由表4可知,UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法测定结果与国标方法的结果基本一致,无显著性差异,但前者所需时间更短,精密度更好。[/size][align=center][size=16px]表4 奶粉质控样品[color=black]BQC1051147452[/color]生物素的测定结果[/size][/align][table][tr][td][align=center][size=16px]检测方法[/size][/align][/td][td][align=center][size=16px]特性值区间(ug/100g)[/size][/align][/td][td][align=center][size=16px]测定平均值(n=6)[/size][/align][/td][td][align=center][size=16px]相对标准偏差/%(n=6)[/size][/align][/td][/tr][tr][td][size=16px]UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法微生物法[sub] [/sub][/size][/td][td][align=center][size=16px]15.6~22.4[/size][/align][align=center][size=16px]15.6~22.4[/size][/align][size=16px][/size][/td][td][size=16px]18.718.1[/size][/td][td][align=center][size=16px]2.5[/size][/align][size=16px] 4.6[/size][/td][/tr][/table][size=16px]3 结语本文建立了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)测定奶粉中[color=black]生物素[/color]含量的分析方法。该方法具有较高的灵敏度、准确度和精密度,前处理步骤简单,分析速度快,特别适合大批量样品的检测。参考文献:[1] GB 5009.259-2016 食品安全国家标准 食品中生物素的测定.[2] 薛霞, 赵慧男, 魏莉莉, 等. 超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中五种水溶性维生素的含量[J]. 食品与发酵工业. 2021,47(12) : 250-256.[3] 李佳兴, 周利, 金艳, 等. 超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定枸杞子中8种水溶性维生素[J]. 食品科技. 2018,43(11) : 336-341.[/size]
[table=100%][tr][td]傅立叶红外光谱图~每个峰值都会对应一个特定的官能团吗?与是什么物质有关系吗?没关系的话,从哪可以找到官能团与峰值对应的图谱啊?谢谢谢谢~[/td][/tr][/table]
[color=#333333]质谱定量实验测定乙腈的杂质含量,结果实验结果显示产生CO2??是什么原因,请指教![/color]
SNT 2534-2010 进出口水果和蔬菜制品中展青霉素含量检测方法 液相色谱-质谱 质谱法与高效液相色谱法

[color=#444444]偶尔打样品会峰值变成一簇一簇的的!!!!请问是什么原因?[/color][color=#444444][/color][color=#444444][img=,690,514]http://ng1.17img.cn/bbsfiles/images/2017/09/201709211717_01_2931083_3.png[/img][/color]正常应该是这样子的:[img=,686,548]http://ng1.17img.cn/bbsfiles/images/2017/09/201709211717_02_2931083_3.png[/img]
用液相色谱-质谱对燕窝中唾液酸含量的检测,不知道对流动相的选择有什么要求?与液相色谱法检测选择的流动相有何不同?江湖救急,求各位大神各显神通帮帮忙。
[color=#444444]请问用液相色谱串联质谱法测试水样中农药含量时,为什么要在过滤后的水样中加入等体积的有机溶剂?最终如何计算测得的浓度?是否为2倍关系?[/color]
请问漂移数值设置的大小会对谱图的峰(面积或形状)以及含量造成哪些影响,比如含量(或面积)
最近得到一份岛津的文献方法《气相色谱质谱法测定葡萄酒中104种农残含量》,给大家分享。如附件。没想到葡萄酒也要检测那么多种农残。说明葡萄上打农药也打得很厉害。岂不是很危险,尤其像我这样吃葡萄不吐葡萄皮的。元芳,你怎么看?







 我要推广仪器
我要推广仪器
 下载APP
下载APP