小弟,最近在写英文文章,自己找的英文文章大部分用的是AB的质谱,求用安捷伦6000系列质谱的文章,想知道参数的英文,及仪器描述时的写法。谢谢各位了
各位老师好,我们[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做十六种有机磷,做的0.1标液,然后要把每一项的峰面积都写上,然后一百多份报告要抄100多遍,感觉工程量不少啊,我觉得我们这种写法太繁琐,各位老师有简单的写法么?[img]http://ng1.17img.cn/bbsfiles/images/2018/05/201805271208134184_5537_1848108_3.jpeg[/img]
怎么样才能得到质谱图上单个质谱峰的丰度?我用agilent MSD chemstation 分析GC-MS数据,求帮帮忙,
一点思路也没有,特来请教各位。反应液样品,色谱有两个峰想知道对应的质谱图是怎么样的,但是质谱不出峰,猜测这两个峰对应的物质质谱响应应该会有,就是比较弱而已,质谱状态正常,扫描范围足够大。用了各种流动相,具体有:1、甲醇/乙腈+0.1%甲酸水 2、甲醇/乙腈+8mmol/L乙酸铵水 3、甲醇/乙腈+8mmol/L乙酸胺0.08%甲酸水 完全没有任何响应,按照猜想那峰的浓度估计得有数十个ppm了,实验室只配了esi源,无APCI源。接下来还有什么处理思路吗?
电喷雾质谱切完石英毛细管后,分子质量不准了,而且质谱峰跟色谱峰一样,是中空的,显示的质量基本不是样品的分子量,请各位指点,我们的质朴是fenigen公司的LTQ-XL型
[color=#444444]我现在的化合物是一个酸性化合物,还有一个羧基,所以做了一个负离子的质谱,C12H13NO3 (M-1):219.24,理论上是218.24 但是质谱图上是218.93[/color][color=#444444]还有个也是一样的,有一个羧基,结构类似,同样做的负离子峰[/color][color=#444444]C6H9NO3 (M-1):143.14, 理论上是142.14但是质谱图上的峰是142.81[/color][color=#444444]我后面做了核磁,核磁上也是貌似对上了。就想问问大家,这个质谱有没有可能是这样子的啊?大家要是有相关文献最好啦。。。[/color]
急求各位大侠。bruker q-tof(micro TOF-Q III)仪器直接进样寻找反应中间体。一级质谱看到159和169的峰,选取169作为母体峰继续二级质谱。问题是: 二级质谱中始终能看到159的峰(而且是簇峰),难道是母体峰169可以继续反应?
如题,之前求助过质谱出倒峰的问题,后来发现是由于柱子的问题,请问柱子为什么会导致质谱出倒峰呢?另外,质谱出现倒的小毛刺正常吗?
我用水和乙腈做流动相,做液质时,第一针是好的(就有一点点质谱峰,是242),第二针质谱图后面,质谱基线一直往上飘,然后水平,基线一段内都是242峰,用95乙腈冲半小时都有242,用低浓度乙腈冲,242渐渐就没有了,这是什么原因呢?(梯度是0-17分钟29%-74%乙腈,,然后74%等度10分钟),242峰从16分钟渐渐出来
超好的论文摘要写法
前段时间ABI的质谱公司赵贵平经理在讲座时提到不怕 几个峰叠到一起就怕峰型不好质谱可以把几个物质在同一时间峰分开并举列子说有36个物质在一个峰图里,但是只有27个 峰(36和27为大概意思)可以分为四个图来说明有36个物质的峰大家有了解这些的么 有什么资料么?
用SIR扫面某个离子,TIC有个小峰,但是点开这个峰的质谱图却看不到任何一个质量数的峰存在,这是为什么呢?附件中其他的峰都有质谱图,为什么4.23的峰没有质谱图?
LCMS中,流动相为ACN和水,双相加TFA,质谱信号会出现M+H,2M+H等峰,会出现M+H+ACN的峰吗?液质中会出现峰吗?会经常出现吗?一般在什么情况下会出现峰啊?
[color=#444444]有个问题要问大家:[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]时为什么在液相中可以出峰,而在质谱上怎么打一级质谱反而没峰出来啊?我是ESI源[/color]
[color=#444444]做一个三萜类化合物,发现接枝一个基团后,做质谱出现了一个M+88的峰,我是柱层析后直接做的质谱,洗脱液中含有乙酸乙酯。不知道这个东西是不对还是结合了一个乙酸乙酯分子。[/color][color=#444444] 还有我做了一个R-N3(叠氮)化物的质谱,里面出现了一个M峰,因为叠氮就是一个正离子,那么在电喷雾质谱中是不是直接就出现M峰呢??希望各位大侠能给指点一二,万分感谢。。[/color]
怎么样才能得到质谱图上单个质谱峰的丰度?我用agilent MSD chemstation 分析GC-MS数据,求帮帮忙,» 引用回复
我有一个质谱图,化合物精确分子量为522.0266.。理论上的M+1 峰应该是 522.0300+1.0078=523.0378。现在正离子模式 质谱图上出现了522.0312(丰度100)和523.0270(丰度35)问题:图谱上523.0270应该是M+1峰,看丰度不应该是同位素造成的而是仪器造成的,问522.0312的峰如何解释,同一个质谱会同时出现M和M+1吗?
如何判断质谱图目标峰,什么是假阳性
质谱棒状图出现一堆峰,堆积在一块形成一根很宽的棒状图是怎么回事啊?
做淫羊藿苷的质谱分析,液相DAD都出峰了,质谱不出峰,采用的文献的乙腈-醋酸水的方法,采用正离子模式检测,郁闷
液相出峰,但接上质谱后就不出峰瓦里安的液质,对溴苯腈,用的乙腈/水做流动相和配样。我在只有液相时能走出峰,但是接上质谱后就没有峰,全是噪音。不是一个样的问题,我做过好几个样都是这样。我是新手,麻烦大家帮我下,在这先谢谢了。
目前在做质谱参数优化,4个化合物,均为卤代甲苯类化合物。仪器为Agilent 6470QQQ,scan模式下正负离子扫描,化合物紫外吸收很好,但其TIC图均未出现明显峰形,提取EIC图也没有峰。同事说该类化合物很难离子化,后试过将水相改为0.1%FA溶液及10mmol/L乙酸铵溶液,促进离子化。只有一个物质在10mmol/L乙酸铵溶液条件下得到加钠峰的EIC图,但与其对应的紫外吸收峰时间间隔了1min,样品从紫外直接流入质谱检测,一般情况下0.2-0.3min就可以完成,因此对此存在质疑。大家碰到过这种情况吗?或是有什么建议吗?
质谱调谐峰校正错误,工程师更换了固定透镜的螺丝,(期间拆过预四级杆,四极杆,检测器),装后调谐通过,一天后做样发现所有碎片和分子离子峰均成了M-1,再调谐无法通过,提示峰校正错误。
各位老师,很多人说利用DAD判断的峰纯度不可信,推荐利用质谱来判断。请问质谱怎么判断峰纯度呢??能否举例说明下,谢谢!
[table=100%][tr][td]我用agilent MSD chemstation 分析GC-MS数据,怎么样才能得到质谱图上单个质谱峰的丰度?[/td][/tr][/table]
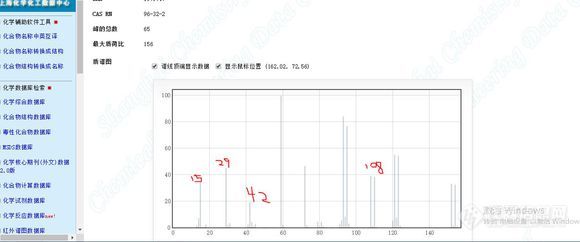
[b]乙酸甲酯质谱15、29、42、108的峰是怎么出来的,[color=#333333]想了半天没想出来它这几个峰的断裂反应,求大神把这几个峰的断裂过程或形成原因告诉我。[/color][color=#333333][img=,580,242]https://ng1.17img.cn/bbsfiles/images/2019/05/201905081121164214_2264_1823055_3.jpg!w580x242.jpg[/img][/color][/b]
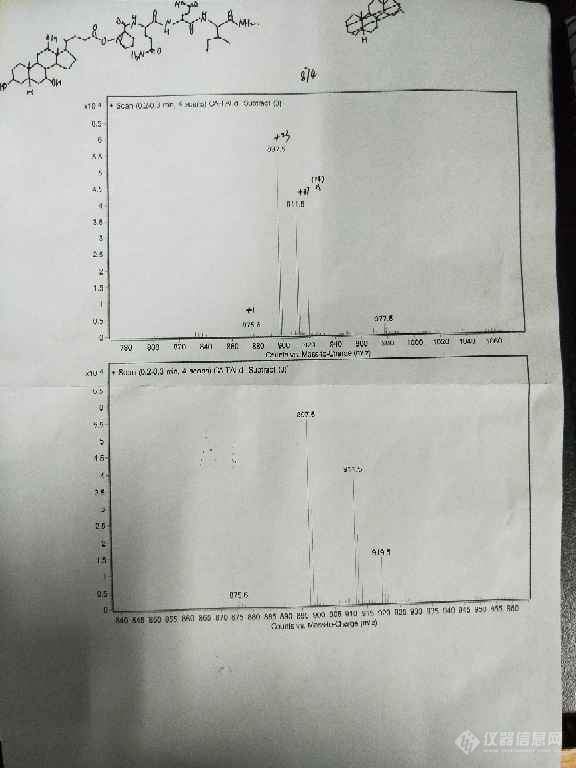
[color=#444444]第一次接触质谱的小白,想请教一下我产物打出来的质谱右边两个峰是怎么回事,我的化合物分子量大约874左右,ESI源,甲醇作溶剂[/color][color=#444444][img=,576,768]https://ng1.17img.cn/bbsfiles/images/2019/05/201905081405365109_7970_1849104_3.jpg!w576x768.jpg[/img][/color]
LC-MS做出的两种谱图是什么呢在别的实验室做了一个LC-MS得到两种谱图,但不知道专业的名称,比如一种是质谱检测到的HPLC谱图,另一种是紫外检测器检测到的HPLC谱图,下面分别对应每条色谱峰的质谱图。谢谢大家不吝赐教!
在做多环芳烃检测时,(之前柱子刚做过抗氧化剂),在8min左右出现小峰,在23min左右出现一个10的7次方的峰,打开质谱看从50到290全是粒子碎片,而且基本上是按照14的质量数递增的,不知道这个是什么,不是我的目标物,现在已做了以下调整1,换隔垫,2,检查衬管不脏,石英棉不黑,只有一点隔垫碎屑;3.切柱子。4,换空白溶剂,走甲醇,乙腈,丙酮都存在相同的峰,走刚开封乙腈这个峰低些,走空针和水没有大的峰,8min左右的小峰一直都有。但这些调整都没有使峰消失。求各位大神帮忙找一下这是哪里的问题,谢谢,质谱图在附件。
气相质谱测定物质过程中,如果目的峰附近有一个较大的峰,这两个峰可以基本达到基线分离,那么这个大峰有没有可能会造成目的峰定量不准啊?







 我要推广仪器
我要推广仪器
 下载APP
下载APP