请问您在没有质谱情况下,用过双色谱柱定性定量吗?
我们单位最近想进一台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],主要用来检测农残,根据农业部的标准,要求是双柱定性检测,以前没接触过这个方法,有没有哪位了解的,给指点一下!!
在使用GC测定农残时,假阳性和假阴性是不可避免会出现的,对与这两种情况的确证,可以用质谱定性,也可以用双柱定性。大家平时都是怎么选择的?我们实验室都是用质谱确定的,现在挣考虑使用双柱法,希望大家指导一下哪个使用更为方便和简单,谢谢!
一直开开心心简简单单的做着有机磷的操作,保留时间简简单单就好。结果临时上任操作有机氯,操作没问题。想问一下如果一个标准物质在[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的谱图上用双塔双柱检测,前后柱子都是各四个峰。可是在检测实际样品的时候,前柱子出现其中两个峰,后柱子上也出现两个峰,这样的谱图可以定性定量么?亦或者是前柱子出现两个峰,后柱子出现三个峰,又该怎么处理?
请教双柱双气路结构的色谱,是不是进样口、柱子、检测器都要配两个,那做实验时两个进样口,难道还要各进一次样吗,好多都是双柱配置,谁有这方面的仪器资料
我在书上看到,在定性分析中可靠性最大、最可以让人接受的是双柱分析,即使用两支固定相不同的色谱柱各分析一次,保留时间如果都相符合,就可以确认。问题来了我用的是HP6890,我分析了无水乙醇和异丙醇,我发现在程序升温和恒温两种条件下两种物质的出峰时间是不同的,那么同一柱子在不同条件下的结果都不同,如果是不同的柱子如何能得到相同的保留时间?

【科学仪器DIY】一机多用: 在一台气相色谱质谱联用仪上面实现双进样口、两根色谱柱自动切换;同时使用MS和FID双检测器的尝试Simultaneously Application ofDual-Column and Dual Detectors on Chromatograph-Mass Spectrometry-FID System摘要:应用双进样口、双毛细管色谱柱以及质谱检测器(MSD)和氢火焰检测器(FID),通过四通分流装置在一台带FID的气相色谱质谱联用仪(GCMS)上面实现一根毛细管柱同时采集MSD和FID双检测器信号数据;同时安装两根不同极性的柱子,不需要拆装柱子,进行无痕切换使用来满足不同样品分析要求。关键词:气相色谱质谱联用;氢火焰检测器, 双柱子双通道Abstract: MSD and FID are simultaneously applied in dual inlet, dual column and GasChromatograph-Mass Spectrometry with FID(GC/MS/FID)with 4-way splitter. It is not needsto dismount or mount different column to satisfied different analysis needs.Keywords: GCMS; FID, dual detector; dual column.前言有时候实验室可能会遇到一台仪器多种功能同时使用或一机多用的要求。原来自己的实验室仅有一台GCMS,一般安装极性毛细管柱子,主要测定挥发性香气组分,但有时候也需要非极性毛细管柱子来测定一些样品。如果更换柱子,就可能要关机,抽真空平衡等,换柱子总有点麻烦,感觉不方便,也浪费时间。另外由于MS的定量线性范围小,对于复杂的天然香气组分的样品的分析很不方便,所以一般采用MS定性,FID定量。这是由于FID的动态线性范围很宽,香气香味样品一般用气相色谱FID来定量,而不用质谱总离子(TIC)来定量 。即需要解决两个问题:一是MS和FID同时测定,二是一台仪器同时能安装两根不同极性的柱子且最好能无痕切换以适应不同的分析要求。从文献看,一般测定复杂香气化合物样品多是用GC-MS的TIC定性,GC-FID来定量(一般需要两台仪器),也有用TIC定量。1试验部分1.1 仪器与装置美国安捷伦6890N/5975C气相色谱-质谱联用仪,带有德国Gerstel的MPS2多功能自动进样系统,德国Gerstel的CIS4大体积分流/[fo
请问神马叫“双柱定性”?哪位老师有相关学习资料?
气相色谱与有双柱箱的吗?这样一台就能当两台用呀!
可利用气相色谱仪,采用火焰离子化检测器(FID)和色谱柱,并使用给出的气相色谱测试条件,分别记录校准化合物在两根色谱柱(所选择的两根柱子的极性差别应尽可能大)上的色谱图;在相同的色谱测试条件下,对被测试样做出色谱图后对比定性。为什么要两根柱子取去对比定性?谢谢了。
卤代酚卤代酚是含酚的卤代化合物,对革兰氏阳性菌有强杀菌作用,用在化妆品中的卤代酚有六氯酚 等多种化合物。这类化合物通常是光敏物质。我国化妆品卫生标准规定为限用物质,限用量见表2-3-17。表 2-3-17 化妆品卫生标准中卤代酚的限用量品名 序号 最大使用量(%) 溴氯双酚 4-4 0.1 双氯酚 4-7 0.2 2,4-二氯二甲苯酚 4-8 0.1 三氯生 4-21 0.3 六氯酚 4-24 0.1 4-溴邻甲苯酚 4-31 0.3 苄氯酚 4-42 0.2 4-氯2-甲苯酚 4-55 0.2 4-氯3,5-二甲苯酚 4-56 0.2 * 指化妆品卫生标准(GB7916-87)中的序号,4-42即表4的序号42(一)薄层色谱法(TLC)1 适用范围本方法适用于化妆品中六氯酚,双二氯酚硫醚、二氯酚和三溴水杨酞替苯胺的定性。2 原理样品经预处理后,样液中的卤代酚用的薄层色谱法进行分离、呈色,然后与标准斑点比较,进行定性。3 试剂3.1 乙醇:分析纯。3.2 己烷:分析纯。 3.3 丙酮:分析纯。3.4 无水硫酸钠:分析纯。3. 5硫酸(lmol/L)。3.6 六氯酚标准溶液(1):准确称取用苯重结晶的六氯酚50.0mg,加丙酮溶解后移入50ml容量瓶中并定容至刻度,避光保存。此溶液1ml含1.0mg六氯酚。3.7 双二氯酚硫醚(2):准确称取用苯重结晶的双二氯酚硫醚50.omg,用丙酮溶解,移入50ml容量瓶中并定容至刻度,避光保存。此溶液lml含1.0mg二氯酚硫醚。3.8双氯酚标准溶液(3):准确称取用甲苯重结晶的双氯酚50.0mg,用丙酮溶解,移入50ml容量瓶中,定容至刻度。此溶液1.0ml含1.0mg二氯酚,避光保存。3.9三溴水杨酞替苯胺(4):准确称取用丙酮重结晶的三溴水杨酞替苯胺50.0mg,用丙酮溶解,移入50ml容量瓶中并定容至刻度。此溶液1.0ml含1.0mg三溴水杨酞替苯胺,避光保存。3.10 乙醇一己烷(1 9)。3.ll离子交换纤维素(5):将DEAE(二乙基氨基乙醇)纤维素,(交换量约0.9meg/g),浸泡于50倍量的0.lmol/L的盐酸中,用玻璃漏斗过滤,用20倍量的丙酮,30倍量的0.lmo1/L氢氧化钠溶液淋洗至OH-型后,用水洗成中性,再用20倍量的丙酮淋洗,弃去丙酮。空气中干燥。保存在乙醇十己烷(l+9)溶液中。3.12 硅胶:薄层用硅胶中加有荧光剂。3.13碱性氧化铝。3.14展开剂:石油醚 冰乙酸(89 12)3.15显色剂。3.15.1 浓氨水。3.15.2 2%4-氨基安替比林溶液:称取2g4-氨基安替比林用乙醇溶解稀释至100ml。3.15.3 8%铁氰化钾溶液(K3[Fe(CN)6])。3.15.4 2%三氯化铁溶液(FeCl36H20):称取2g三氯化铁用乙醇溶解稀释至100ml。3.15.5 2%铁氰化钾溶液。3.16 盐酸 丙酮溶液:9.5ml盐酸加丙酮至100ml(临用前配制)。4 仪器 4.1 层析柱:、内径10mm、高200mm的具塞玻璃管的下端熔接玻璃过滤器或塞有玻璃棉,4.2 紫外灯,具有8W功率,254nm波长。4.3离子交换柱(6):将离子交换纤维素用乙醇 已烷(3.11)配成混悬液,:用湿式填充法缓慢倾入层析柱中,以防止产生气泡,填充高度80mm。5 分析步骤5.1样品预处理(7)(8)称取含卤化酚0.5mg的样品(扑粉,除臭砂芯、香波约10g,膏霜约0.5g),置于100ml玻璃瓶中,连接好回流冷凝器:加50ml乙醇 已烷(1 9)溶液,2ml 1mol/L硫酸,于水浴上加热3min,冷却后用3号玻璃砂芯漏斗过滤,用乙醇 己烷(1 9)溶液5ml洗沉淀,滤液移入分液漏斗中静置分层。取己烷层用10ml水洗涤,无水硫酸钠脱水后以0.5ml/min的流速注入离子交换柱(10)。用50ml己烷洗涤。去除油脂等干扰物质,弃去淋洗液,依次用10ml丙酮、2ml丙酮 盐酸溶液(3.16),20ml丙酮洗脱(11)。溶出液在水溶上加热蒸去有机溶媒,加5ml乙醇,加热使盐酸挥发,重复此操作2次。残渣加2.0ml丙酮溶解,作为样品待测溶液。5.2 制备薄层板5.2.1硅胶薄层板:硅胶30g,加水约65ml,搅拌均匀,涂布成厚度0.25~0.3mm的薄层板,105~l10℃干燥30min,置干燥器中保存。 5.2.2含硝酸银的氧化铝薄层板:0.12g AgNO3,加少量水溶解,加30ml乙醇、20g氧化铝,调成浆状物,涂布厚度为0.25~O.3mm的薄层板,空气中干燥、于干燥器中避光保存。5.3 点样距薄层板底边2cm处将5~20μl待测溶液从左到右点样(12),两点间隔约1cm,薄员板.的右边点2μl标准溶液,空气中干燥。5.4 展开取适量展开剂(3.14)倾人展开槽中,将薄层板放入展开剂中,待溶剂上升约10cm,取出薄层板,空气中干燥。5.5显色(13)在薄层板上顺序喷雾显色剂3.15.1~3.15.3或3.15.4~3.15.5,六氯酚在显色剂3.15.1~3.15.3中为红色,在3.15.4~3.15.5中为蓝色斑点。二氯酚硫醚和三溴水杨酞替苯胺在3.15.1~3.15.5中为紫色,在3.15.4~3.15.5中呈现蓝色斑点。用加荧光剂的硅胶薄层板测定时,各种卤代酚在紫外线照射下,在各自的Rf 值位置上以荧光为背景呈现出暗黑色的斑点。
硫双威原药该产品有效成分硫双威的结构式和基本物化参数如下:化学名称:3,7,9,13-四甲基-5,11-二氧杂-2,8,14-三噻-4,7,9,12-四-氮杂十五烷-3,12-二烯-6,10-二酮结构式http://ng1.17img.cn/bbsfiles/images/2014/12/201412271243_529409_2960432_3.png 实验式:C10H18N4O4S3相对分子质量:354.5(按2007年国际相对原子质量计)生物活性:杀虫熔点:(173~174)℃蒸气压(20℃):5.1Pa溶解度:水中35mg/L,丙酮中8g/kg,甲醇中5g/kg,二甲苯中3g/kg。稳定性:60℃稳定,其水悬浮液因日光而分解,pH6稳定,pH9迅速水解,pH3缓慢水解。 1 范围本标准适用于硫双威及其生产中产生的杂质组成的硫双威原药。2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 1600-2001 农药水分的测定方法GB/T 1601-1993 农药pH值的测定方法GB/T 1604-1995 商品农药验收规则GB/T 1605-2001 商品农药采样方法GB 3796-2006 农药包装规则GB/T 6682-2008 分析实验室用水规格和试验方法GB/T 19138-2003 农药丙酮不溶物的测定方法HG 2611-1994 灭多威原药3 要求3.1 外观:白色至类白色固体。3.2硫双威原药应符合表1要求。表1 硫双威原药控制项目指标 项 目指 标硫双威质量分数,% ≥95.0灭多威质量分数,% ≤0.5水分质量分数,% ≤1.0pH值4.0~7.0丙酮不溶物质量分数,% ≤0.54 试验方法除另有说明,本试验所使用的试剂均为分析纯试剂,水应符合GB/T6682-2008中的三级水规格。4.1 抽样按照GB/T1605-2001中“商品原药采样”进行,用随机法确定抽样的包装件;最终抽样量不少于100g。4.2 鉴别试验 本鉴别试验可与硫双威含量的测定同时进行。在相同的色谱操作条件下,试样中某一色谱峰的保留时间与标样溶液中硫双威色谱峰的保留时间,其相对差值应在1.5%以内。4.3 硫双威质量分数的测定4.3.1方法提要试样用50%(V/V)四氢呋喃甲醇溶液溶解,以甲醇+水为流动相,使用Kroasil C18为填充物的不锈钢柱和可调波长紫外检测器,对试样中的硫双威进行高效液相色谱分离和测定。4.3.2试剂和溶液 水:新蒸二次蒸馏水,经0.45μm滤膜过滤;甲醇:色谱纯;四氢呋喃:HPLC; 硫双威标样:已知含量,≥99.0%。4.3.3仪器高效液相色谱仪:具可变波长紫外检测器;色谱数据处理机;色谱桩:250mm×4.6mm(id)不锈钢柱,内填Kroasil C18[
使用双柱色谱测量乙酯含量时,双柱的成分,柱前压是否应完全一样?请专家指导。
[font=宋体][color=black][back=white]定性方法:用已知保留值定性;根据不同柱温下的保留值定性;根据同系物保留值的规律关系定性;双柱、多柱定性;双检测器定性或利用检测器的选择性定性;利用其他物理方法结合定性,如质谱;利用化学反应或物理吸附作用对样品进行预处理。[/back][/color][/font] [font=宋体][color=black][back=white]定量方法:外标法;内标法;叠加法;归一化法。[/back][/color][/font]
气相色谱单柱单气路和双柱双气路有什么区别?我不明白什么是双柱双气路?
[color=#444444]操作一台带有双检测器和双进样口的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],无论从气路的连接和软件都有点无从下手,比如两根柱子都接上的话,是不是一根柱子不用载气也得开着,还有什么是需要开着的呢,还有应该注意些什么呢,跪求各位有经验的大神指点一二[/color]
安捷伦双泵1260上有个像色谱柱一样的东西,但不是色谱柱,它的作用是什么啊
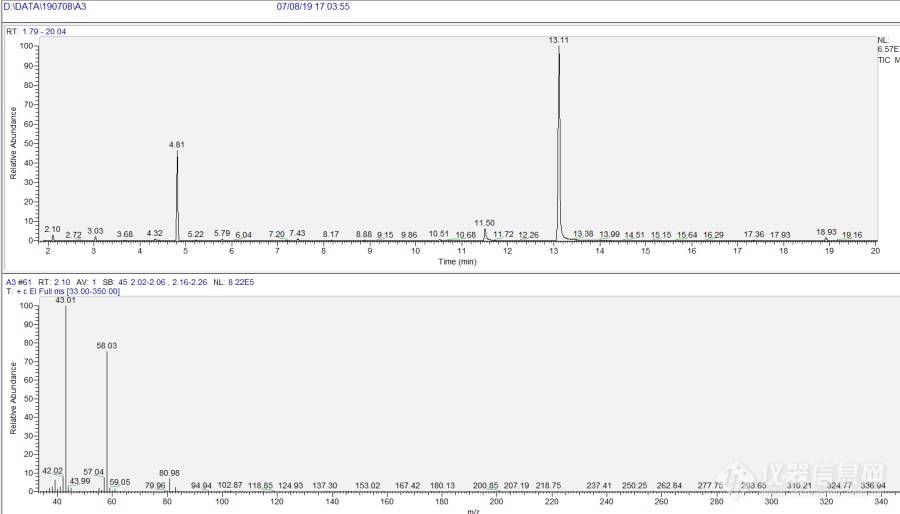
7月接到一样品:AB胶中A胶,要求[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS定性其中挥发性溶剂。经过一段时间摸索定性出其中10种溶剂,现把过程写出来供交流,指正。 首先定性未知物要尽可能了解样品信息,经过交流得知样品是AB胶中A胶,为无色粘稠液体,组成为环氧树脂加稀释剂,稀释剂可能为苯甲醇。要求定性其中挥发性溶剂至少要知道主要的溶剂。因为含树脂不能直接进样,查了下苯甲醇沸点206℃,公司没有自动顶空连接[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS,所以考虑手动顶空 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS进样。 样品前处理:取样品10ml于20ml顶空瓶加盖密封,微量注射器也同时放入烘箱在85℃恒温30min。 色谱质谱条件:[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS为 Trace1300-ISQ 色谱柱 TG-5MS (30m*0.25mm*0.25μm)35℃以4℃/min升温至260℃,柱流速1ml/min,顶空进样量250μl,分流比20 ISQ 全扫描33-350 溶剂延迟1.9min[img=,690,394]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171413539863_4070_2103464_3.png!w690x394.jpg[/img][img=,690,339]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171429246599_2108_2103464_3.png!w690x339.jpg[/img]经过NIST检索 2.10min S-环氧丙烷;3.03min 1,2-二氯乙烷;4.32min 1,2-丙二醇;4.81min 甲苯;10.51min 苯甲醛;11.50min碳酸丙烯酯;13.11min 苯甲醇;18.93min正十二烷。这里2.10min峰 乍一看有熟悉的43,58 以为是丙酮,检索出来却是S-环氧丙烷。由于保留时间过小保留指数参考意义不大,于是取丙酮(分析纯)蒸汽进样看看保留时间:[img=,690,359]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171425533408_1516_2103464_3.png!w690x359.jpg[/img]丙酮保留时间2.07min 和2.10min很相近 所以通过保留时间无法判断是不是丙酮。考虑到最低扫描是从33开始扫描丢失了低端碎片,于是改条件从14开始扫描,扫到150,同时考虑到质谱图比检索出的S-环氧丙烷多了81,83峰,这个检索出的S-环氧丙烷还可靠吗?是不是有同流出呢?于是降低初始温度 为30℃保持10min 以10℃/min升温至260℃,柱流速0.7ml/min尽量分开低沸点组分,溶剂延迟1min以免漏检。[img=,690,391]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171445320329_6041_2103464_3.png!w690x391.jpg[/img]前面大峰是空气峰,因为强度大有分叉,2.64min 检索得S-环氧丙烷(正匹配884,逆匹配947),81,83峰(较小)还是形影不离。这81,83峰是什么?困扰了好久还发贴问了。后来我觉得应该是共流出,申请买了一根 Stabliwax-MS (30m*0.25mm*0.25μm)柱,40℃保持2min 以4℃/min升温至250℃,柱流量1ml/min 分流比20 ISQ全扫描14-150 顶空进样250μl:[img=,690,391]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171455520831_8615_2103464_3.png!w690x391.jpg[/img]在1.85min出来的峰检索得到 S-环氧丙烷 而且没有 81,83碎片!确定之前那个81,83就是共流出了,那么这81,83究竟是何方神圣? 通过离子监测发现1.70min有它们的踪迹,峰极小,检索得到1-氟-1,1-二氯乙烷,原来81,83峰是痕量的1-氟-1,1-二氯乙烷 干扰所致。[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171609285159_9183_2103464_3.png!w690x388.jpg[/img][img=,690,355]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171459047641_5780_2103464_3.png!w690x355.jpg[/img]极性柱 NIST检索的结果是 1.85min S-环氧丙烷;2.88min 甲醇;5.29min 甲苯;5.65min 水;6.02min 1,2-二氯乙烷;9.64min 正十二烷;19.79min 苯甲醛;21.86min 1,2-丙二醇;28.93min 碳酸丙烯酯;29.73min 苯甲醇比较下两柱定性结果,极性柱多了 甲醇和水,因为在TG-5MS 柱这两个都随着死时间出峰 所以没检测到。另外这个水峰 会不会是空气里带来的呢,因为顶空瓶在空气里加,空气本身的水分呢,于是在极性柱打个空白看看[img=,690,390]https://ng1.17img.cn/bbsfiles/images/2019/08/201908171512104229_628_2103464_3.png!w690x390.jpg[/img]发现空气空白TIC图也有水峰,但比样品显著少些,于是判断样品确实有水分。空白无其他干扰峰。结论:1.通过双柱手动顶空进样 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS分析得到A胶中的挥发性溶剂有: S-环氧丙烷,甲醇, 甲苯,水, 1,2-二氯乙烷, 正十二烷, 苯甲醛, 1,2-丙二醇, 碳酸丙烯酯, 苯甲醇。其中主要溶剂是苯甲醇。 2.有共流出的情况,双柱定性是个很好的选择,两根柱子的极性尽可能差别大些。 3.极性柱对于低分子量极性组分分析更有优势。 4.未知物定性全扫描范围尽可能的低些以免丢失低端碎片。
做农残分析时,我知道两个柱子定性是为了双确认。那么定量呐,不知道用那个柱子定量做有机磷是FPD 检测器A柱:百分之五十聚苯基甲基硅氧烷DB-17柱直径0.53MM。 B柱:百分之百聚甲基硅氧烷DB-1 直径 0.53MM有机氯 ECD 检测器 A柱:百分之百聚甲基硅氧烷DB-1 直径 0.25MM B柱:百分之五十聚苯基甲基硅氧烷DB-17柱直径0.25MM我知道DB-1 好像是非极性的。请大哥大姐赐教哈~~ 想知道的确切些~~http://simg.instrument.com.cn/bbs/images/default/em09512.gif还有DB-17 和DB-1701 区别
第三方检测实验室对于测苯系物 和TVOC 两方面来说,气象色谱仪是选择一台具有双填充柱、双进样器和采样器、双气路系统结构的,还是选择 两台单进样器单采样器单填充柱的气象色谱比较好?有的说选择两台比较好,可是选择具有双填充柱、双进样器和采样器、双气路系统结构的不更简洁方便吗??不太懂 求解。
双FID检测器双填充柱进样口的国产[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]多少钱
如何考察色谱柱的稳定性?自己装的c18色谱柱 如何考察其稳定性?谢谢
小白持有上海精科GC112A,配备TCD检测器、六通阀进样器、双色谱柱①Parapak Q柱+②GDX-104柱载气为氢、流量4.1ml/min、检测器120℃、进样器70℃、柱温40℃。原先采用进样口1(对应Parapak Q柱)对甲烷标准气体检测,甲烷峰和氩峰(平衡气)不能完全分离。现在采用进样口2(对应GDX-104柱)进行检测,却不能出峰,这是为什么啊~~~(ps:以前该仪器的进样口2没用过的~~)
SunFire色谱柱 最好的低PH值稳定性 最佳粒度和批次重现性 满足制药行业最严格的性能要求下载SunFire色谱柱介绍资料(PDF) http://www.china-hercules.com/ziliao/sunfire.pdfSunFire 是一个崭新的、代表先进技术的反相 C18 键合硅胶色谱柱品牌,它提供优异的峰形、出色的低 pH 稳定性以及杰出的柱效。不仅如此,更大的样品容量与独特的优化柱床密度 (OBD) 设计的结合,使 SunFire 制备柱具有无与伦比的性能和制备放大能力。对于制药行业的苛刻要求,SunFire 色谱柱的性能绝对优于市场上具有竞争力的所有主流 HPLC 硅胶色谱柱。SunFire 分析色谱柱 - HPLC分析色谱柱分离性能的新纪元优异的峰形SunFire 色谱柱综合了最新的键合技术和端基封尾技术,提供对称的峰形,能够在低、中pH条件下对酸性、中性和碱性组分得到更好的分离度和柱效。杰出的柱效SunFire 色谱柱使用先进的硅胶填料,并应用最新的C18键合/端基封尾技术专利以获得杰出的柱效,同时也提高了灵敏度。出色的低pH 稳定性优越的低pH稳定性 (超过市场上绝大多数硅胶色谱柱品牌) 延长了色谱柱寿命。MS兼容性SunFire色谱柱完全适于质谱应用,同时拥有所有的优点—优异的峰形、极佳的灵敏度、更大的峰容量和极低的柱流失。无与伦比的批间重现性SunFire色谱柱的制作过程严格控制,以确保达到最高级别的柱间、批间色谱重现性。SunFire 制备OBD 色谱柱 - 制备色谱柱性能的新纪元优化柱床密度 (OBD) 设计优化柱床密度设计 (OBD) 是一个耗时2年的研发项目最新成果。 这项技术综合了机械因素、填料特性和填料柱床密度因素,最后得到了拥有优秀的稳定性、卓越的重现性、超高的上样量和极高柱效的全新色谱柱。超常的柱寿命对于纯化实验室来说,无论是高通量样品使用周期还是制定年度预算计划,柱使用寿命都是一个值得考虑的关键性因素。 SunFire 制备 OBD 柱具有优化的柱效和超乎寻常的柱寿命 (超过 1000 次DMSO进样)。良好的柱效SunFire 制备 OBD 色谱柱提供从分析到制备级别相同的色谱性能,消除了方法重新开发的需求。超高的样品容量SunFire 制备硅胶填料特殊的合成技术确保在酸性和中性pH下能达到最大的样品容量。精确的制备放大能力SunFire 制备填料和色谱柱的生产环境完全符合 cGMP 要求,填料合成和柱装填过程均有最严格的质量控制,以确保精确的制备放大能力。全国免费服务热线:400-716-3003
各位大虾,您们好!您们哪家的GC是双柱双通道来做样品的初步定性和定量的,两条色谱柱的前端是由一个切换器来转换的,它们转换的时间那个品牌的GC会更快点???我们这边是做血液中乙醇含量的测试的
各位版友,我现在有一个问题,如果采用双柱双FID来检测血液中酒精的含量,但样品就一个,是顶空的时候重复进样还是怎么弄?如果是重复进样的话,第二次进样是否就不准确了,只能辅助定性,而只能采用第一次进样来做定量分析。另外还有一种方式就是一个进样口采用三通分流道两根色谱柱上面,但准确性怎么样?谢谢!
RCONH22 确定初始操作条件主要包括进样口温度、检测器温度、色谱柱温度和载气流速。分流进样的进样量一般不超过2μL ,最好控制在 0 .5 μL 以下 ,进样量还和分流比有关 ,分流比大时 ,进样量可大一些 ;进样口温度应接近或高于样品中最重组分的沸点 ;对于一个未知的新样品, 可将进样口温度设置为 300 ℃;常用毛细管GC 所用柱内载气线流速为:氮气 20~40 cm/s。隔垫吹扫设定为 2 ~5 mL/min , 分流比依据样品情况(如待测组分浓度等)、进样量大小和分析要求来改变, 选择一个合适的折衷分流比,用分流比范围 20∶1 ~200∶1 ,待测组分浓度大或进样量大时, 分流比可相应增大,反之则减小,用大口径柱时分流比小一些,用微型柱做快速GC 时,分流比要求很大,流比小时, 分流歧视效应可能小,但初始谱带(主要是溶剂谱带)宽度大,分流比大时,初始谱带(主要是溶剂谱带)宽度小,但分流歧视效应可能大。检测器温度可参照色谱柱的最高温度设定,而不必优化。色谱柱温度,组成简单的样品最好用恒温分析;组成复杂的样品,常需要用程序升温分离;色谱柱的初始温度应接近样品中最轻组分的沸点, 最终温度取决于最重组分沸点;升温速率依样品的复杂程度而定,建议毛细管柱的尝试温度条件设置为OV -1或SE-54 柱 :从 50 ~280 ℃,升温速率 10 ℃/min ,V - 17(OV -1701)柱:从60 ~260 ℃, 升温速率 8 ℃/ min ,PEG -20M 柱:从60 ~200 ℃,升温速率 8 ℃/ min 。这是方法开发时的初始参考条件,具体工作中再根据样品的实际分离情况来优化设定。3 尝试性分析上述初始条件设定后,便可以进行样品的尝试性分析。一般先分离标准样品,然后分析实际样品。在此过程中,还要根据分离情况不断进行优化。GC的分离优化就是要在保证分离度和灵敏度的前提下,实现快速分析。在实际工作中,一般是首先满足分离度的要求,然后提高分析灵敏度,最后再考虑尽可能缩短分析时间。改变柱温和载气流速可改变分离度;内径越小,或者填料粒度越小,柱效越高;薄液膜色谱柱的柱效高于厚液膜柱;更换色谱柱可改变分离度;用化学作用如通过生化反应改变待测物结构;程序升温是GC分离复杂混合物的有效方法;进样量小一些、进样口温度高一些、载器气流速快一些、汽化室体积小一些,分流比大一些,对窄的初始谱带宽度有利。4 气相色谱定性与定量分析对于简单的样品,可通过标准物质对照来定性。对于复杂的样品, 则要通过保留指数定性和或GC/MS来定性。对于基层监测站,气相色谱定性分析最主要是依据保留值定性,即在相同的条件下,分别注入标准样品和实际样品,根据保留值确定色谱图上哪个峰是要分析的组分。但必须注意,在同一根色谱柱上,不同的化合物可能有相同的保留值,对未知样品的定性仅仅用一个保留值还不够。双柱或多柱保留指数定性是气相色谱定性分析较为可靠的方法,不同的化合物在不同色谱柱上具有相同保留值的几率要小的多。建议对复杂的样品采用双柱或多柱保留指数法定性。气相色谱定量方法包括面积百分比法、归一化法、外标法、内标法、标准加入法。基层监测站最常用的方法是外标法,只要用一系列浓度的标准样品做出工作曲线, 就可以在完全一致的条件下对未知样品进行定量
请教下:色谱柱柱温太高对柱子的稳定性的影响?最近试着的高温柱温下进行样品分析,柱温升温达到较高温度时,谱图基线严重不稳,经常出现一些鬼峰,且样品分析的平行性很低(特别是在做低含量样品分析时,很困扰)。觉得问题是高温下柱子固定相不稳定,柱流失严重。是不是? 还可能会有什么原因呢?有什么解决方面呢?本人现用色谱柱DB-1301,柱温升到260+温度后,基线很不稳定。
[center]色谱定性分析(一)利用纯物质对照定性在一定色谱条件下,一个未知物只有一个确定的保留时间。因此将已知纯物质在相同的色谱条件下的保留时间与未知物的保留时间进行比较,就可定性鉴定未知物。若二者相同,则未知物可能是已知的纯物质;不同,则未知物就不是该纯物质。纯物质对照法定性只适用于组分性质已有所了解,组成比较简单,且有纯物质的未知物。 (二)相对保留值法相对保留值是指组分i与基准物s调整保留值的比值,即: 它仅随固定液及柱温变化而改变,与其它操作条件无关。相对保留值的测定方法:在某一固定相及柱温下,分别测出组分i和基准物质s的调整保留值,再按公式可求出的值。 (三)加入已知物增加峰高法当未知样品中组分较多,所得色谱过密,用上述方法不易辨认时,或仅作未知样品指定项目分析时均可用此法。首先作出未知样品的色谱图,然后在未知样品中加入某已知物,又得一色谱图。峰高的增加的组分即可能为这种已知物。 (四)保留指数定性法保留指数(retention index) I 是柯瓦(Kovats)于1958年所提出的,所以又称柯瓦指数。它表示物质在固定液上的保留值行为,是目前使用最广泛并被国际上公认的定性指针。它具有重现性好、标准统一及温度系数小等优点。保留指数的物理意义在于:它是与被测物质具有相同调整保留时间的假想的正构烷烃的碳数乘以100。保留指数仅与固定相的性质、柱温有关,与其它实验条件无关。其准确度和重现性都很好。 (五)其它方法 除利用保留值定性外,尚可利用检测的选择性响应的特点将未知物大致分类,还可与化学反应结合起来,是一种简便、有效的定性方法。此外,还可与别的方法,如红外光谱法(IR)、质谱法(MS)、核磁共振波谱法(NMR)等结合进行定性鉴定。[/center]
双气路色谱仪器,厂家分别安装了填充柱和毛细管色谱柱?同时安装使用,有冲突吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP