色谱分析定量方法为归一法,分析结果如何以算术平均值报出?
请问安捷伦1260-6400质谱定量软件中单点校正和面积归一化方法怎么设置?谢谢!
是这样的,质谱例如乙苯的定量离子是91/106,那么我看质谱图的时候发现它的碎片是91.1/106.1,再进一针发现它的碎片是91/106,在做线的时候选择的定量离子是91.1/106.1,这样是对于91/106是可以正常定量的吗,会对结果有什么影响吗,例如定不了量之类的,感谢老师??
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]可以用面积归一化法来定量吗?
请教各位专家:我用的是四级质谱,想根据特征峰用它进行定量。但是我发现在同一条件下,纯物质的各个碎片峰比例不是定值,比如说噻吩,它的两个比较大的峰是m/z84和58,可是测定的过程中,两个峰的强度比值在不同的时间不相同,请问这是什么原因啊?如果比值不定我就无法用特征峰进行定量。多谢
三重四级杆质谱是如何定量的?三重四级杆通过离子打碎获得特异性子离子,子离子在通过Q3后时在接收器上转化为电信号,反映到分析软件就是离子强度图。在一定线性范围下,分析物浓度越高,打到接收器上的离子就越多,信号越强,这是定量的基础。此外在定量时可以选择用峰高或峰面积定量,一般选用峰面积定量准确。因为是定量,所以必需标准曲线,原理与高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]类似。一般多用内标法定量,以排除质谱重现性和样品处理造成的影响。那么,随着设置离子对越多,如何能保证定量准确呢?四极杆对于离子的选择性通过是交替进行的,在所有离子通道之间快速切换。随着设置离子对数量的增加,每个离子所占用的检测时间缩短,这会影响到其检测灵敏度,但是只要实际样品和标准溶液所采用的分析方法一致,定量的准确性理论上是不受影响的。
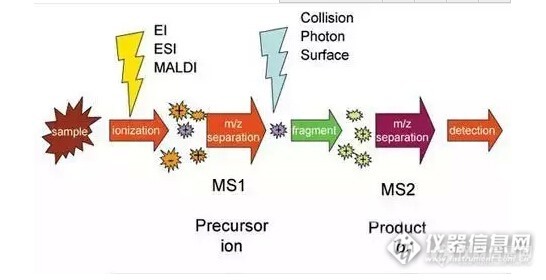
质谱信号。与EI谱图分析以相对强度为主不同,在色谱-质谱联用时,信号的绝对强度就成了我们天天都要关心的内容,因为质谱信号强度随时间的变化就是实验的色谱图,通常以总离子强度或者某一特定质荷比离子的强度作图。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271813_575350_2544766_3.jpg2、定量的两种方法外标法 用已知量的标准样品A和未知量的待测样品A分别进行实验;我们会得到以下三个信息:标准样品的量(已知);标准样品的信号强度;待测样品的信号强度。(假设样品的响应=常数*浓度,从这三个信息即可算出待测样品的量。) 为了更加精确地测定未知量的样品,我们希望标准样品的信号强度与待测样品的信号强度尽量接近(以减少非线性响应的影响)。因此常用的外标法会测量一系列已知量的标准样品,绘制一条工作曲线,再用拟合的方法确定未知样的量。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271814_575351_2544766_3.jpg内标法 外标法主要有以下两方面的局限:1标样和待测样是独立进行实验的,实验间的偶然误差无法消除;2标样和待测样的基质(即除待分析物外的其它成分)不同,基质有可能会带来不同的影响,也会产生误差。 那么,如果我们把已知量的标准样品B直接加入待测样品A,就可以把标准样品和未知样品的测定在同一次实验和同样基质中完成,也就消除了两次实验和基质不同造成的误差,这就是内标法。(如果加入的标准样品和待测样品是同种物质A,那么由于它们不可区分,只通过一次实验是不能定量待测样的,这时我们在加入标样前后分别进行两次测量,即测量待测样及待测样+标样的信号,即可计算出待测样的量。)3、质谱相关的特殊定量细节同位素稀释 前面内标法的介绍中我们可以发现,最理想的内标物既要和待测样相同(具有相同的响应系数)又要不同(仪器可以区分二者的信号),这对矛盾的集合体就是同位素内标。 由于不同同位素的化合物具有近似相同的物理化学性质,离子化时的响应通常也是相同的,而它们具有不同的质荷比m/z,即可在质谱中被区分出来。因此同位素标准品是最理想的内标物。 另外,由于某些元素的天然同位素分布有一定的比例,当我们加入一定量的同位素内标时,可以把对信号绝对强度的测量转化为对信号相对比例的测量,从而提高实验的准确性。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271814_575353_2544766_3.jpg选择反应监测 在不太复杂的体系中,我们只要按照分子量就可以定性某种化合物了。但对于复杂混合物(如石油产品/生物样品)而言,很多化合物具有相同或相近的质量(同分异构体质量完全相同,有些化合物分子量非常接近,如CO和N2,要考虑仪器的质量分辨率是否能区分二者),此时仅靠测量质量就不能确定这个化合物是否就是我们关心的“the one”了。 在串联质谱 (Tandem MS) 仪器中,我们不仅可以把质谱仪理解为一个称量离子的“天平”,它还具有了离子“镊子”(选择某个特定的离子把它分离出来)和“剪刀”(把某个/某些离子激活并打成碎片)的功能。通过母离子和子离子的两步选择,我们可以在复杂体系中精确定位到我们关心的化合物,同时,两次离子选择还可减少复杂基质的干扰,降低背景噪声(获得更低的检出限)并提高方法的动态范围。因此选择反应监测是目前色谱(气相色谱/液相色谱)-质谱联用中最常用的定量方法。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271815_575354_2544766_3.jpg选择反应监测在不太复杂的体系中,我们只要按照分子量就可以定性某种化合物了。但对于复杂混合物(如石油产品/生物样品)而言,很多化合物具有相同或相近的质量(同分异构体质量完全相同,有些化合物分子量非常接近,如CO和N2,要考虑仪器的质量分辨率是否能区分二者),此时仅靠测量质量就不能确定这个化合物是否就是我们关心的“the one”了。在串联质谱 (Tandem MS) 仪器中,我们不仅可以把质谱仪理解为一个称量离子的“天平”,它还具有了离子“镊子”(选择某个特定的离子把它分离出来)和“剪刀”(把某个/某些离子激活并打成碎片)的功能。通过母离子和子离子的两步选择,我们可以在复杂体系中精确定位到我们关心的化合物,同时,两次离子选择还可减少复杂基质的干扰,降低背景噪声(获得更低的检出限)并提高方法的动态范围。因此选择反应监测是目前色谱(气相色谱/液相色谱)-质谱联用中最常用的定量方法。
你好,如果我采用质谱定量,用内标法,内标的含量保持恒定,按比例增加标准样品的浓度,以标准样品和内标的比值及标准样品的浓度做标准曲线,会不会因为标样的浓度越来越大,产生离子抑制,而使标准曲线趋势降低,结果偏低?
我用的是热电的GC-MS,对于软件的操作不是很熟悉。由于我是刚接触对于质谱的定量问题一直不是很清楚,看了一些书,介绍的方法和单纯气谱的差不多。所以,想请各位高手指点一下,如何利用质谱图进行定量分析呀!谢谢各位的帮忙!
我之前定量没有用同位素内标,保留时间也能分开。最近看到关于质谱定量内标选择的内容,认为同位素内标好,能校正基质效应,色谱行为和响应特征接近,即便没有合适的同位素内标,也要选择结构类似的,色谱行为接近的。但是如果液相分不开一起进质谱的话,会不会产生离子抑制?
各位老师,大家好,新学质谱定量分析,请问质谱定量要用哪一种模式呢?我们做的一级质谱的信噪比很低啊,还不及紫外,定量为什么要用到二级质谱呢?烦请各位老师赐教。
质谱的应用越来越广泛,良好的定性能力是大家公认的,但是说到定量,很多人都认为质谱的定量结果准确性不如色谱,真的是这样吗?发表一下您的看法吧。好的回帖另有加分,技术版面,请勿灌水!
如题,请教各位大神,如何理解高分辨质谱“一级定量和二级定性”呢?
关于质谱图中的定量离子与定性离子的确定方法:定量离子是不是以质谱图中最高的相对离子强度为定量离子,定性离子的选择则以特征离子的丰度比为选择依据啊,我是初学者,多谢指教
中国古代《矛和盾》讲述了一个人自称拥有最坚固的盾和最锋利的矛的故事,于是出现了自相矛盾的笑谈。质谱,作为日益普及的检测工具,已经被广大检测人员所熟知。多数人会有这样的认识,三种四级杆是用来定量的,飞行时间是用来定性的。就目前发展而言,某些新出的飞行时间质谱会宣称除了具有超强的定性能力,也同时具备了三重四级杆的强的定量能力。这是否也是自相矛盾呢?同样的,大家有没有考虑过:为什么三重四级杆质谱仪的发展现在比较慢了?是不是遇到了什么瓶颈为什么大部分质谱公司都不能推出具有能测定准确分析质量的四级杆呢?你是如何理解质谱的定性与定量之间的关系的?是鱼和熊掌不可兼得、自相矛盾吗?以上仅代表个人观点,欢迎版友拍砖!http://simg.instrument.com.cn/bbs/images/brow/em09504.gif
顶空固相微萃取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱联用测定挥发性物质 如何定量?
http://www.instrument.com.cn/bbs/shtml/20060406/385022/我把它弄成附件的形式发出来[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=107513]影响质谱定量参数[/url]对于“影响质谱定量参数”的学习心得,抛砖引玉先!!![color=#DC143C][B]本帖子结合“影响质谱定量参数”资料和具体的试验操作的学习心得[/B][/color]资料中指出影响质谱定量参数的7个大因素:1、组成色谱峰的点 至少要在15-20之间,才能保证峰的稳定性,增加点的方式有:提高扫描速度,但是这样可能会导致检测限的下降。 [color=#DC143C]我想作为方法建立者,只要基本上按照仪器提供的最优值就应该没有问题,虽然对我们影响不大,但我们至少要知道有这么回事,呵呵;[/color]2、 离子喷雾的稳定性 离子源喷雾不稳定影响方法的优越性是显而易见的,流动相混合的不好、过长时间的流动相、喷雾毛细管切口不好、聚酰胺突出均会导致该结果,切割倒是很重要的,必须切正。[color=#DC143C]我在使用中好像流动相是经过严格超声的,喷雾哪个地方的毛细管因为有时系统压力太大需要切割,但一般是切割的地方都是顺着过来的那端,没有动过喷雾口的那段,聚酰胺突出的情况也没有怎么去留意。[/color]3、峰形就不说了,主要是会影响到积分的效果,峰形好的定量更好,峰形差选择积分点很难导致不稳定。4、色谱分离 尽量将各个不同的物质分开,避免相互之间的抑制。[color=#DC143C]这点倒有值得斟酌,如果几个物质同时出,因为都是微量的,难道它们就不能都远远的达到被完全电离的程度吗?我想肯定是可以的,这也是质谱的优势嘛,尽量分开都说是可以提高灵敏度的。大家也来发表一下看法哈[/color]5、流动相中添加物 要严格按照规则来,要使用挥发性的盐,如醋酸铵、甲酸铵等,尽量不使用TFA,尽量不使用离子对试剂。6、内标的使用 可以减少每次操作中的误差,减小方法的变异系数,它说的是需要内标与该非同位素的药物分开,而我做试验过程中也使用到了内标,它们两者是很难分开的,保留时间基本一样,还好的是母离子,子离子都不一样,都相差相同的数字。如果遇到子离子相似的情况是不是像它说的那样呢,“The labeled standard should be “far” enough away from the non-labeled to avoid signal contribution of the abundance of the natural isotopes to the signal of the internal standard”,大家也来讨论一下啊!!!7、质谱的分辨率 这个很重要,我们使用的TSQ好像是低分辨率质谱,不知道什么是高分辨率的质谱???呵呵
看到一篇文献,介绍[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]分析尼古丁时,使用QQQ可以比高分辨质谱获得更低的定量限,请问这种说法是否正确?
背景:多晶硅含碳偏高,主要是三氯氢硅精馏提纯不够。企业一般是热氢化的回收氢和还原炉的回收氢混着用,氢化炉在1300度以上时,它的石墨罩会和氢气生成CH4,含量高的时候能达到几百个PPM,在还原炉中分解沉积在硅棒中。还有就是吸附塔再生时也能产生一定量的CH4。大家都知道甲烷热裂生成炭黑和氢气的反应,就是隔绝空气加强热到1000至1500度左右.在1100度以下时不可能出现石墨与氢气化合生成甲烷的反应,或者大家也可以用热力学计算一下,先假使二者可以反应,通过平衡常数计算出的含量能有几个ppm就很不容易了。 CH4(g)=C(石墨)+2H2(g) 反应自发温度823K多晶硅生产中氯硅烷中总碳(甲基硅烷)质谱分析?
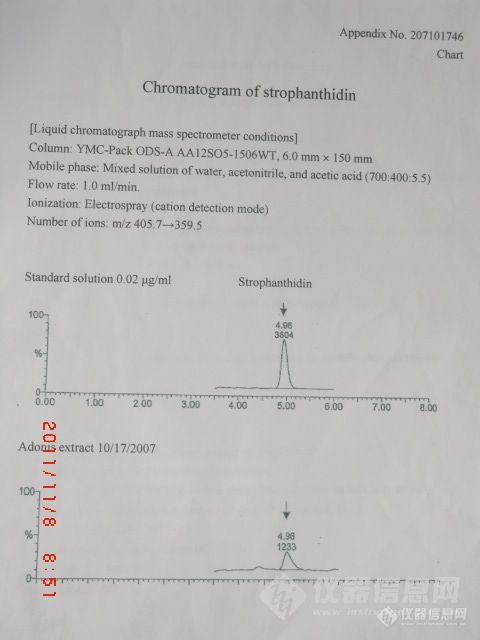
大家好,请教大家个问题。由于偶没用过质谱,对下面的检测条件及其谱图看不懂,这个图是直接用质谱定量的还是先用质谱定性后又用液相定量的?平时做的液相图,没发现纵坐标是这种形式的,令俺很费解。还有,我的标准品配成0.2ug/ml在液相上的检测效果就不太好(安捷伦的),但这个材料中,浓度比我的减小了十倍,效果还非常好,这是什么原因?难道真的是用了质谱定量,其定量效果真的比液相好?请帮忙解答,不胜感激。http://ng1.17img.cn/bbsfiles/images/2011/11/201111080908_328971_2404619_3.jpg
想请教下,质谱和色谱定量的差别?
1 要用目标离子的碎片定量,特征性强,排除干扰;2 在定量分析的方法设置上,尽可能提高扫描速率,提高准确率和重复性(可以通过a减小扫描质量数的范围来提高目标峰的扫描次数,或 将一个样品全部分析时间断分成n个segments,对目标离子单独设置扫描模式);3 一定要通过色谱柱分离后定量分析,避免竞争性离子的存在影响目标离子的离子化效率;如果目标分子未与竞争性分子完全分开,则在离子化过程中导致目标分子的离子化效率降低,导致样品分子的定量结果偏低,当然标准浓度的样品也要用相同的方法分析。4 如果样品都是纯品的话可以不经过色谱柱直接进样分析,包括做标准曲线的样品(虽然不建议直接进样分析)。5 如果用的离子源的喷针位置是可移的话,一定要记住做标准曲线时其位置,否则其位置移动后在相同的条件下进入质谱的离子流量会发生改变,标准曲线就不能使用了,白忙!对于调用的质谱方法不要改动shealth gas and aux gas 的流速,否则会影响进入质谱的样品量(谢谢Esquire提醒) 6 所建立的标准曲线一个月后如果想重复使用,则用QC样品检验一下该标准曲线!7 对于已经建立好的分析方法在扫描范围、流动相的组成、梯度或流速等方面不要作任何改动,否则,标准曲线要重作。扫描范围改变目标峰的扫描次数、流动相组成改变离子化效率,流速改变色谱峰的保留时间和峰宽。8 离子阱的强项在于多级-定性,四级杆的强项在于定量;9 对于热稳定性不好的样品可以通过提高气速,降低毛细管温度的方法保证定量分析的重复性;一旦方法固定后不要轻易改动;10 仅供参考,欢迎探讨!!
目前在使用HP-5MS(30*0.25*0.25),对同一个样品进行分析,柱流量为1.0mL/min,分流比分别设定为30:1和60:1,其他色谱条件保持一致,且进样量同为0.2uL。那么现在问题来了,对一个样品进行分析,出来的质谱图基本相同(相同的物质都能在对应的出峰时间出峰,只是峰面积大小的问题),但其中有部分含量较少的组分,在两次不同分流比进样的面积归一化法结果中含量竟然相差一倍(分流比为30:1的含量约为3.3%,分流比60:1的含量约为1.7%),究竟是什么原因呢?是分流歧视吗?
[color=#444444]质谱是四极杆检测器。请问其定量方法是怎样的?[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]时[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]定的是峰面积,那么质谱定量定的是什么?怎么定的。求详解[/color]
质谱的定量效果如何?
我用质谱做样时,最后定量出来匹配度很低,我把时间范围度小点匹配度就高了,是怎么回事?
请问各位专家,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱分析农药残留做定性定量分析时,怎么选择定性和定量离子?依据是什么?谁能分享一下相关的资料!谢谢!
谁能提供一些有关质谱定量分析的经验?谢谢!!

我们用的是FID检测器,毛细管柱,用面积归一化定量,可是最近的谱图分离效果跟以前不太一样,所以对仪器的自动积分仪也产生怀疑,不知道其定量是否准确。附谱图一张,请教一下大家看看这样积分是否正确,如果不正确需要如何修改,恳请各位指点一下。http://ng1.17img.cn/bbsfiles/images/2011/10/201110092024_322481_1621482_3.jpg
熊陈思慧等建立了超高效液相色谱_线性离子阱/静电场轨道阱高分辨质谱法同时测定化妆品中的22种功效成分,样品采用甲醇超声提取C18 色谱柱 (100mm *2.1 mm,1.8μm)分离,以0.1% (v/v)甲酸水溶液和乙腈为流动相进行梯度洗脱 在正离子模式下以保留时间和一级母离子精确质量数进行定量分析以高能碰撞诱导解离获得的二级碎片离子精确质量数进行确证结果表明该方法线性关系良好检出限LOD 为 0.003~2.01mg/kg,定量限LOQ 为 0.02~4.36mg/kg水乳霜 种基质中 个添加水平的回收率范围为63.2%~125.1%相对标准偏差为0.18%~10.9%对标示含有烟酰胺抗坏血酸葡糖苷咖啡因泛醇及甘草类光果甘草根茎叶甘草根胀果、甘草根、麦冬根人参根黄芪根虎杖根苦参根地黄根积雪草茶叶提取物的54批样品进行检测标示单体功效成分的样品均有检出标示不同植物提取物的46批样品中24批检出植物提取物的功效成分,该方法简便快速定性定量可靠适用于化妆品中22种功效成分的定量测定。 文章具体内容见附件







 我要推广仪器
我要推广仪器
 下载APP
下载APP