现用的64位ridasoft win酶标仪数据处理软件不显示拐点数量,怎么办?
ICP中每峰点数按照定义来说是定量测试被分析谱线中的峰形需要用几个象速点,最终数值将按照平均值进行,范围是1-7,通常谱线在250nm以下,可以选择2-3-个点,谱线在250nm以上,建议选1个点,ICP中每峰点数大家如何如何理解?
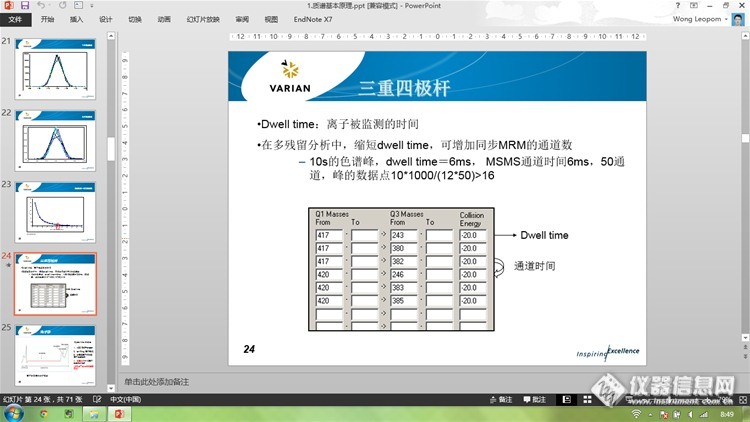
[font=宋体]为了得到较好的色谱峰型,一般要求一个色谱峰应包含[/font][font='Calibri','sans-serif']12-20[/font][font=宋体]个采集点。质谱方法中所设置的一次循环时间,为质谱采集一个点的时间,即循环时间[/font][font='Calibri','sans-serif']=[/font][font=宋体]色谱峰峰宽[/font][font='Calibri','sans-serif']/[/font][font=宋体]采集点数。同时,质谱在一段时间内的循环时间,为这段时间内所有检测通道的驻留时间(该通道下离子被监测的时间)、扫描间隔、空片段时间之和,即[/font][font=宋体]循环时间[/font][font='Calibri','sans-serif']=[/font][font=宋体](驻留时间[/font][font='Calibri','sans-serif']+[/font][font=宋体]扫描间隔[/font][font='Calibri','sans-serif']+[/font][font=宋体]空片段时间)[/font][font='Calibri','sans-serif']×[/font][font=宋体]通道数。因此,一般情况下,色谱峰峰宽为[/font][font='Calibri','sans-serif']0.2-0.3min[/font][font=宋体],要求循环时间控制在[/font][font='Calibri','sans-serif']0.8s[/font][font=宋体]内。[/font]
如题,得到的谱图峰的点数太少了(7个左右),它与采样频率,DWELL time,还有sanpling rate(SCAN),resolution的选择(high or low)有什么关系吗?如果点数要多的话怎么设置?再请教个问题:当SCAN和SIM同时用的时候,他们之间的时间是怎么分配的?一个一半?
标准品跑出来的图很漂亮,纵坐标都是0.0几,但是我配的供试品出来的峰很扁,纵坐标大一个数量级,峰面积也不小了,就是峰很不容易看出来,请问这是怎么回事呀?[img]https://ng1.17img.cn/bbsfiles/images/2020/12/202012010941081910_5585_5082352_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/12/202012010941081724_3874_5082352_3.png[/img]
如何增加[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]样品检测数量?
如题,接触液质时间不长,很多问题搞不清楚,如题所述2002/657/EC中表6是关于质谱技术和联用技术获得识别点数,其中离子数一列有用N表示的,鉴别点数一列有用n表示的,在这里N和n分别表示什么意义?还请大家帮忙。2002/657/EC的文件我已上传,还请大家帮忙。http://ng1.17img.cn/bbsfiles/images/2013/06/201306032229_442830_1644952_3.bmp
作为经常摆弄仪器的“行内人”,大家在做实验的同时,或多或少也有着对色谱峰的审美,也就是通常大家说的,这峰出的“不好看”,今天咱们就来聊聊这气相色谱峰的身材问题,怎么才能做出“好看”的峰形呢? 理想情况下,经色谱分离的峰应该为高斯分布曲线,即对称峰。但实际上当一个样品谱带沿着色谱柱前进时,由于浓度差等原因,样品分子会向谱带两侧扩散,从而使色谱柱出口处的样品谱带比柱入口处宽,且可能产生不对称的峰,就是谱带展宽。谱带展宽的程度主要用柱效来表示,色谱峰越对称,峰越窄,柱效越高。 影响谱带展宽的因素有多种,主要分为柱内和柱外两种。柱内因素是指色谱柱本身的性能,如柱活性大小、固定相是否与样品发生化学反应、柱效是否够高、样品是否超载等等。柱内因素导致谱带展宽的因素则主要是指街头的死体积、进样口和检测器死体积等。在气相色谱中,柱外因素导致谱带展宽的程度要比柱内因素小得多。色谱峰除了会变大、变宽,向圆头峰、平头峰的出现也时有发生,下面我们就来逐一聊聊。峰面积变大 导致色谱峰峰面积变大的主要原因有以下几点:①方法参数设置:a. 分析条件的改变,如环境温度升高,分流比变化等。分流比变小,峰面积变大。b. 数据处理机问题。比如重新开机后,可能会出现这种情况;c. 方法设置参数变化,如积分参数变化;d.手动进样时进样技术不好。②仪器因素:a.载气流速控制不好,流速增大或柱前压力调节阀异常,可能出现这种情况;b. 分流口被污染;c.程序升温过程中升温重复性不好,柱温控制不良;③色谱柱因素:a. 色谱柱类型不适合分析该样品,导致固定液流失;b. 柱温过高超过了色谱柱固定液的温度上限,导致固定液流失;柱温太靠近色谱固定液的温度下限,导致样品在流动相和固定相之间的分配比发生变化;c. 色谱柱用了段时间,未老化,柱性能变差甚至有之前的样品残留物,可能导致这样情况;d.柱温未达到平衡就开始进样; 解决方案 测定时色谱峰面积变大的现象,在确认没有改变色谱条件和方法参数的前提下,首先要考虑进样问题。如果是手动进样,要提高进样技术,进样量要准确、稳定。如果是自动进样,则维护、改善进样系统,保证进样器正常工作;其次,考察仪器因素。要观察载气压力是否稳定、柱前压调节阀是否有问题;测定分流口和隔垫吹扫口排出的载气,排出气是否减少,必要时调整分流比或清洗分流口。另外,要记录升温过程(柱温,如有程序升温的进样口也要考察)的温度变化,控温精度是否正常;柱温的控制是否正常尤其重要。如有问题,需要维修温控系统的电路部分。 当然,色谱柱的因素也必须考虑。色谱柱的固定液类型是否适合分离该样品,柱温是否合适等。如果色谱柱不合适,更换色谱柱。如过色谱柱用了很久,没老化,就老化后再做;如果老化色谱柱,柱性能仍不能恢复,那么得更换新的色谱柱。平头峰 色谱图中出现平头峰,首先要考虑进样量是否过大,导致信号过大,信号超过记录仪的最大测量值,不再上升出现的平头峰,包括进样量过大及浓度太大等;还可能是检测器灵敏度选择太高,离子化检测器所用静电计输入达到饱和,记录仪滑线电阻或机械部分故障。 解决方案一旦遇见平头峰,应该从以下来解决:①减少进样量,或对样品进行合理稀释,或进样时加大分流比;②适当调节检测器信号衰减,改变记录仪量程;③增大色谱仪上衰减倍数,减小灵敏度。当然,在色谱分析时,定量的依据是色谱峰响应大小与组分量在一定范围内呈线性关系。对有些试验而言,主要考察的是杂质量,所以有时为了能准确检测出有关杂质化合物的量,往往会通过增大供试品溶液浓度,提高杂质的信号响应值来进行试验,这时供试品主峰就可能会出现平头峰,因杂质峰的响应值与主成分峰的响应值相差太大且无关,因此不必关注此类主成分平头峰,对杂质首选自身标准品对照定量法,实际试验中要注意具体问题具体分析!圆头峰色谱分析中出现圆头峰,有以下几个方面原因:①进样量过大,超过检测器的线性范围(ECD检测时尤其如此);②检测器受固定相流失及样品中高沸点成分、易分解组分及腐蚀性物质的污染;③记录仪灵敏度过低;④载气系统可能存在泄漏。 解决方案针对色谱图中出现圆头峰,可采取的措施有:①减少样品溶液进样量或将样品稀释后再进样,或增大分流比来进样;②清洗检测器,如果污染物仅限于高沸点物质,则通常可将检测器加热至最高使用温度后,再通入载气就可清除,要注意加热的温度不能损坏检测器的绝缘材料;如果加热法不适宜,也可以用丙酮等溶剂从进样口注入(每次可注入几十微升)进行清洗,在污染程度较轻时是有效的;若以上方法都不能解决污染问题,则应将检测器卸下,选择既能溶解污染物又不损坏检测器的溶剂,用注射器注入测量池进行彻底清洗;③适当调节记录仪灵敏度;④查看载气气路压力,仔细检查是否存在泄漏,这种情况一般伴随着保留时间或响应值的变化。
最近打液相色谱老是出现一些杂七烂八的峰,而我要的色谱峰却不出现,因此我想可能是色谱柱已经坏了,才导致出峰有问题,我是一菜鸟,不能确定。所以我想请问各位如何判定色谱柱如何判定已坏?
[b][font=宋体]问题描述:按照[/font]HJ 786-2014[font=宋体]测定土壤和沉积物中多环芳烃,色谱条件:岛津[/font]LC-20AT[font=宋体]四元低压液相系统,二极管阵列检测器([/font]SPD-M20A[font=宋体])和荧光检测器([/font]RF-20A[font=宋体]),流动相为乙腈水溶液,流速为[/font]1mL/min[font=宋体]。结果出峰数量少两个并且与标准中多环芳烃的出峰顺序不同,这是为什么?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])现有流动相洗脱条件无法使所有目标物完全分离,可设置不同的梯度洗脱程序,观察单峰是否有分离成两个峰的迹象,并且验证峰纯度,如条件允许,也可使用质谱或购买单标进行定性。[/font][font=宋体]([/font]2[font=宋体])借助单标或者质谱验证,确认是否混标中本身就缺少某种组分,或是某种组分已经分解或是损失。如确认是标准品本身的原因,应重新购置合格的标准品。[/font][font=宋体]([/font]3[font=宋体])尽量采用推荐的色谱柱,液相色谱法测试[/font]16[font=宋体]种多环芳烃有专门的色谱柱,可以使用的安捷伦公司和岛津公司的[/font]PAH[font=宋体]专用柱,规格是[/font]4.6mm×250mm[font=宋体],[/font]5μm[font=宋体]。[/font][font=宋体]([/font]4[font=宋体])分离效果与仪器的延迟体积也有一定关系,比如岛津的[/font]LC-20AT[font=宋体]带四元比例阀加混合器,延迟体积可以大到[/font]3mL[font=宋体]左右,安捷伦的[/font]1260[font=宋体]带四元比例阀延迟体积一般是[/font]900μL[font=宋体]左右;因此同样的梯度、同样的色谱柱分离也不一定完全一样,也会出现某些峰不能完全分离的情况。[/font][font=宋体]([/font]5[font=宋体])出峰顺序与柱子、流动相、柱温等很多因素有关。如果条件不能做到,无法与标准中出峰顺序完全一致,应对色谱峰进行定性确认。如能定性,即便有个别峰顺序不对,结果也是可以接受的,这并不会影响定量分析。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
前沿陡峭,后沿较前沿平缓的不对称峰,称为拖尾峰。气相色谱中,常见的吸附色谱法(利用吸附剂表面对不同组分物理吸附性能的差别而使之分离的色谱法称为吸附色谱法),如果吸附等温线为非线性,当进样试样量超过一定数量时就会出现拖尾峰;分配色谱法(利用固定液对不同组分分配性能的差别而使之分离的色谱法称为分配色谱法),如果载体表面具有活性作用点,试样量超过柱负荷或进样方法不当等,都会出现拖尾峰现象。什么原因会导致色谱峰拖尾?
以前,[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测时,色谱峰面积比较小的还很多,现在,只剩下峰面积很大的,色谱峰数量减少,这是怎么回事?
我用的是Agilent 3000A微型的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url] 在最近机器在开机后进样出现的峰形就是附件中的,色谱仪在稳定半个小时以后峰形才正常。请各位高手支招。
我这几天正在测定蛋黄中FAME,买了sigma的Supelco37FAME混合标准品,[B][size=4]标品图谱的色谱条件为[/size]:[/B]色谱柱:SP-2560,100m*0.25mmID,0.20um柱箱温度:140摄氏度(5min)以4摄氏度/min升至240摄氏度载气:氦气,20cm/sec检测器:FID260进样口:1ul,260摄氏度,100:1[B]我的实验的色谱条件:[/B]色谱柱:安捷伦DB-23,30m*0.25*0.25um柱箱温度:80度3min,以10度/min的速度升至170度,维持10min,再以5度/min的速度升至220度,维持3min载气:氮气0.5ml/min检测器:FID250进样口:1ul,240,20:1由于条件不同,出峰时间与标准品不同,但峰数量远大于37种,近五十种。不知道如何确定各峰与混标中各物质的对应关系,C4-C16确定还相对比较容易,后面就很难了,无论是峰高还是面积与标准品图谱相差很大,是不是我的色谱条件不合适,需要改进?还是有其他的方法?再有标品中浓度是不是和色谱峰面积对应,浓度大峰面积就大?请各位帮忙!
[color=#444444]请教,做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],相同仪器(色谱柱、检测器均相同)做的GC信号,对甲烷浓度的响应系数和对乙烷浓度的系数之间会有数量关系吗?检测器为FID,载气为氮气。真心请教[/color]

RT,我最近需要外送一些样品做拉曼测试,但是填写实验信息时有一栏是让我确认 测试点数,[img=,690,102]https://ng1.17img.cn/bbsfiles/images/2021/08/202108291044448674_2433_5108430_3.png!w690x102.jpg[/img]查了一些文献,然后网上似乎也没找到相关的信息,所以希望求助一下各位....这个测试点数是什么意思,有什么作用呢?
请问MRM采集的点数与采集的离子对数和dewell time 分别是什么关系?麻烦请大神们详细解释下,谢谢
做PBB,PBDE时,绘制标准曲线点数的选择,我是选择1ppm,3 ppm,5ppm绘制曲线,本次实验外审后,专家称点数太少,应该做五点法,点数也不要太集中,我想十溴联苯及联苯醚5点法线性有点难做,响应值偏低,1ppm有时候响应面积才几千,往下再配制0.5ppm,我担心看不到响应面积,由于标准溶液使用的是百灵威的产品,PBDE原始标准溶液为50ppm,因此我配制的最大工作标液浓度最高只能到达5ppm(10种混标PBDE),而且十溴联苯及联苯醚真的不特别容易被玻璃棉吸附,每40针就要更换衬管,请问大家有什么好的建议?分享下
[font=宋体]发帖人:[/font]doxw0323[font=宋体]链接:[/font][url=https://bbs.instrument.com.cn/topic/961852][color=windowtext]https://bbs.instrument.com.cn/topic/961852[/color][/url][b][font=宋体]问题描述:[/font][/b][font=宋体]前沿陡峭、后沿较前沿平缓的不对称峰,称为拖尾峰。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中,常见的吸附色谱法[/font]([font=宋体]利用吸附剂表面对不同组分物理吸附性能的差别,而使之分离的色谱法称为吸附色谱法[/font])[font=宋体],如果吸附等温线为非线性,当进样试样量超过一定数量时就会出现拖尾峰;分配色谱法[/font]([font=宋体]利用固定液对不同组分分配性能的差别,而使之分离的色谱法称为分配色谱法[/font])[font=宋体],如果载体表面具有活性作用点,试样量超过柱负荷或进样方法不当等,都会出现拖尾峰现象。什么原因会导致色谱峰拖尾[/font]?
请问,分散控制系统(DCS)的I/O点数怎么设置,怎么计算?谢谢
请问Scan ,MRM采集的点数是由每个事件的时间决定还是由loop time 决定?
同一样品用两根不同类型色谱柱测试,一个峰多,一个峰少,是不是两组数据包含的样品信息是相同的,只是由于分离效果差异,导致峰的掩盖重叠而峰的数量减少?或者还有别的原因,谢谢!
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]耗材也叫[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]易耗品,如进样口密封垫、进样口衬管、垫圈、喷嘴、管线、接头等,也有把色谱柱、载气等列入其中的。这些耗材种类繁多,其性能极大地影响着色谱系统的运行状况。色谱仪、分析方法的选择固然很重要,但对这些耗材的选择和安装也是不能忽视的,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]耗材有时甚至成为色谱系统运行的瓶颈。 1:气体过滤(净化)器 在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]中,各种气体(载气、燃气、助燃气、吹扫气等)所含的杂质影响仪器的灵敏度和稳定性。 如氧导致固定相氧化,色谱柱损坏,保留值改变;水使部分固定相水解,色谱柱损坏,产生噪声和拖尾;有机化合物或其他杂质会产生噪声和“鬼峰”;另外,颗粒杂质也能使气路控制系统失灵。所以,在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]系统中,气体都必须经过严格的净化。有的系统中安装了过滤器,但处理不当,达不到预期的效果;有的装上过滤器一用就是一年半载,而不管容量是否已经饱和,反成为系统的污染源。常用的过滤器有氧气分析仪器设备、水分和烃类过滤器,将单个的过滤器组合使用,便能有效地去除这些杂质。 过滤器的连接可分为串联式、并联式和混合式3种。串联式是把分别装有分子筛、脱氧剂、活性炭等吸附剂的净化器串联起来,脱水管在前,脱氧管在后;这是最早的过滤器连接方式,其效率低,更换吸附剂不方便。在并联方式中,每个净化器都有一个开关阀,更换其中的一个,不会影响系统的载气运行,使用灵活、方便。混合式是在净化器中装有多种组合的吸附剂,漏气点少,使用方便,效率高。 过滤器安装的位置和数量由使用的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]系统决定。通常采用两种方式,一种安装在气路管线上(气源后),然后气路管线再分至每一台色谱仪;另一种安装在每台色谱仪前,此时,过滤器的位置离色谱仪越近越好。首次安装或更换气源,管路中会混入空气或其他气体,须用冲洗短路接头代替过滤器连接系统来冲洗气路(不连接色谱仪),然后马上装上过滤器。需要特别指出的是,气路管线不能使用塑料管或分析仪器设备橡胶管。 2:色谱柱卡套 色谱柱卡套是连接色谱柱、保证系统密封性的重要易耗品。选择卡套时,要注意卡套使用的温度及色谱柱的直径与卡套内径的匹配,此外,还要考虑检测器的要求。 3:进样口密封垫 不同品种的密封垫,其质量有很大差别,表现特征是各种“鬼峰”的出现。“鬼峰”的数量越少,其峰高越小,密封垫的质量越好。因此,必须选择流失小或基本没有流失的密封垫。 热导检测器的构成 1热敏元件 热敏元件是TCD的感应元件,其阻值随温度变化而改变,它们可以是热敏电阻或热丝。 2.池体 池体是一个内部加工成池腔和孔道的金属体。池材料早期多用铜,因它的热传导性能好,但它防腐性能差。故近年已为不锈钢形式示意图所取代。通常将内部池腔和孔道的总体积称池体积。早期TCD的池体积多为500-800μL,后减小至100-500μL,仍称通常TCD。它适用于填充柱。近年发展分析仪器设备了微TCD,其池体积均在100μL以下,有的达3.5μL,它适用于毛细管柱。 热导检测器的原理 基于不同组分与载气有不同的热导率的原理而工作的热传导检测器。在通过恒定电流以后,钨丝温度升高,其热量经四周的载气分子传递至池壁。当被测组分与载气一起进入热导池时,由于混合气的热导率与纯载气不同(通常是低于载气的热导率),钨丝传向池壁的热量也发生变化,致使钨丝温度发生改变,其电阻也随之改变,进而使电桥输出端产生不平衡电位而作为信号输出。热导检测器是[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法中最早出现和应用最广的检测器。 热导检测器的特点 ⑴热导检测器基本理论,工作原理和响应特征,早在上个世纪六十年代就已成熟。 ⑵由于它对所有的物质都有响应,结构简单,性能可靠,定量准确,价格低廉,经久耐用,又是非破坏型检测器。 ⑶与其它检测器相比,分析仪器设备TCD的灵敏度低,这是影响它应用于环境分析与检测的主要因素
[align=center][font=宋体]如何应对色谱质谱峰 “消失”[/font][/align][align=left][font=宋体]平时逛仪器社区,经常遇到版友咨询“我的色谱峰怎么忽然消失了”、“仪器灵敏度不够吗”、“我只是换了台仪器,同样的方法为什么一点色谱峰都没有了”、“难道是离子对会自己变吗”等等等等。下面也会有很多热心的版主提出自己的经验并附上详细的检查方法,那么我认为遇到这种情况还是要理清思路,比较色谱峰消失前后,自己做过什么操作,是仪器的问题还是自己的操作失误。毕竟每个实验室环境不同,配置也差异很大,那么今天我来分享一下自己在遇到“色谱峰消失”时是如何冷静处理的。[/font][/align][align=left][font=宋体]首先离子对的数量多了,出现错误的离子对很正常,AB需要自己输入离子对,这样失误的几率大大增加,而Thermo质谱的操作就比较人性化,它有在Tune复制,并到Trace Finder建立方法中粘贴的功能,因此我们在遇到色谱峰“消失”时,首先需要检查是否自己的质谱参数被人修改?色谱条件用差了?流动相跑空了?流动相中的盐析出了?这类问题还是挺多的,需要耐心检查。尤其接的是别人的实验任务时,如果出现此类错误,而目标物的真实分子量被自己上网查出并检查是这个原因导致的色谱峰“消失”,是很让人崩溃的。[/font][/align][align=left][font=宋体]再一个就是关注仪器之间的差别,有的实验室仪器多,操作人员也多,经常出现更换仪器做一个项目的事情,这时会出现分子量偏差的问题,但不至于出现色谱峰“消失”,我目前遇见过一次色谱峰“消失”,竟是因为更换的质谱仪负离子模式没有被准确校正,从而导致我的一对负离子目标物“消失”,而我却为了这个问题各种猜测甚至怀疑自己,当被告知仪器的负离子模式在工程师手上很难校正后也是极度崩溃的。[/font][/align][align=left][font=宋体]第三种情况是浓度的错用会出现色谱峰“消失”,明明微克级别的检测,如果错用纳克,那么再浓缩的样品前处理方法也很难检测到,因为我就犯过类似错误。[/font][/align][align=left][font=宋体]最后附上版主的回复,可参考学习。[/font][/align][align=left][font=宋体]“质谱比UV灵敏度高,TIC又没什么选择性,所以基线很高,淹没了你的目标物。考虑吧样品液处理一下,能够用什么方法让你的目标物浓度高一些,其他的东西少一些。[/font][/align][align=left][font=宋体]另外,有的化合物对UV的响应(一定波长下)比质谱的响应好,所以不是所有化合物都特别适合拿质谱来做,虽然它具有很多优点(此处略去N个字)”;[/font][/align][align=left][font=宋体]“是不是源参数设置不合理,化合物高温条件下稳定吗?灵敏度下降还有可能是离子抑制”;[/font][/align][align=left][font=宋体]“说一下可能性吧,第一还是看柱压,出现两次不同的结果,很大原因是柱压的然变化。[/font][/align][align=left][font=宋体]第二,有可能开始进样的时候,洗脱剂只进了一种,虽然从工作站上看不出来,所以最好的方法是平衡基线的时候,用每种展开剂的100%都冲一下”。[/font][/align][align=left][font=宋体]最后感谢仪器信息网提供原创大赛让大家共同交流学习![/font][/align][align=left][font=宋体] [/font][/align][align=left][font=宋体] [/font][/align]
http://ng1.17img.cn/bbsfiles/images/2015/07/201507070848_553858_2733342_3.png
在色谱分析中,检测器因对色谱柱流出的物质产生不同的响应而产生色谱峰,在色谱图中,根据色谱峰来进行定性和定量的分析,所以色谱峰是色谱分析的关键要素,而由于仪器设备和操作及试剂等的原因,色谱峰会出现很多问题;如峰的拖尾、前延、分叉、变形等,峰拖尾最主要的问题。而从3月份的活动帖子来看,大家讨论的问题也基本上是这些,可以先来看看参与活动的帖子1、吃元宵猜灯谜——猜一猜峰前伸的原因2、【峰谜解读】水做谜语让你猜——看图解读畅谈之三:流动相与水3、【峰谜解读】+重叠峰4、【峰谜解读】+分叉峰(你可能想不到的原因)5、【峰谜解读】遇到难题了,是不是色谱坏了(有图)6、【峰谜解读】基线走成这样,也是醉了7、【峰谜解读】GC7820A TCD检测器出现规则的脉冲峰8、【峰谜解读】+这样的色谱图,您见过吗?9、【峰谜解读】基线不平,原因可能有那些10、【峰谜解读】这样的色谱峰有点想不通11、【峰谜解读】GC-2014出现高温区基线紊乱从这些帖子也可以看出,峰前伸、重叠峰、交叉峰、不出峰、峰基线不好等等,依然是大家探讨的问题。可见,这些问题是比较常见的。然后,这些问题的形成,不同的色谱仪器也会有不一样,而且同一个问题的出现,跟我们的色谱条件也有很大关系。以下列出一些色谱版区相关的帖子,供大家参考1、【资料】进样后色谱峰异常的原因http://bbs.instrument.com.cn/shtml/20070611/872407/2、高效液相色谱常见问题分析与对策http://bbs.instrument.com.cn/shtml/20091126/2231326/3、液相色谱版资料汇总之五——HPLC使用、维护、故障分析与处理http://bbs.instrument.com.cn/shtml/20100221/2406689/4、气相色谱常见问题解答http://bbs.instrument.com.cn/shtml/20060304/354091/http://bbs.instrument.com.cn/shtml/20061101/611697/图谱来找茬(第十季)-不规则基线 · 【讨论】图谱来找茬(第九季)-宽峰/馒头峰 · 【讨论】图谱来找茬(第八季)-肩峰/叉峰解析 · 图谱来找茬(第七季)-拖尾/前沿峰解析 · 【讨论】图谱来找茬(第六季)-鬼峰解析 · 【资料】图谱来找茬(第五季)---柱效我来测【板油解析详解】· 【讨论】图谱来找茬(第四季)----钝峰解析 · 【谱图】大家图谱来找茬(第三季)-倒峰解析 · 【谱图】图谱来找茬【第二季】· 【讨论】图谱来找茬【第一季】
各位板油在色谱分析时一般主峰的峰面积多大才合适?也就是进样量和灵敏度是怎么把握的?如果有分流的话,跟分流比也有关系。我的经验是主峰的高不能满偏,也不能太小。一般满偏是大于1000,我要求峰高在700到800之间。你们是怎么考虑这个问题呀?
dwell time是影响色谱峰点数的最主要因素吗?色谱峰点数儿怎么看?点数儿太多了也不好吗?任意一个点的物理意义是什么?
我想请教下如果进空针,总离子流色谱图应该有峰吗?TIC中的丰度应为0还是存在本底?一般进样离子流色谱峰丰度应为多少数量级?还有GC-MS能查看基线吗?我们是agilent6890N-5975gc-ms
本人matlab的一个新手,想用matlab做一个二维色谱峰数量检测程序来分析自己的数据,奈何自己的matlab操作和编码知识实在太落后了,有部分代码处理和操作实在看不明白,在findpeaks函数上看了将近1个多月了,但是还是只看完了1/2,越到后面越难看懂,求哪位在这方面曾经有过研究或正在研究的亲一起交流交流,走过路过的亲们也不要错过啊,findpeaks在各种谱图中应用极为广泛,大家一起把它弄懂了可以有很多好处的啊。对于给在下帮助较多的亲们,在二维色谱峰检测程序编写完成后,源无偿提供源代码共享,在此先谢过亲们了。







 我要推广仪器
我要推广仪器
 下载APP
下载APP