下图为正离子模式下硝苯地平(相对分子质量346)的代谢物质谱图,请问分析离子峰可能是353还是331呢?实验卡在对代谢物进行定性很久了,希望各位老师多多指教![img=,690,490]https://ng1.17img.cn/bbsfiles/images/2023/12/202312061413355858_937_5900538_3.png[/img]
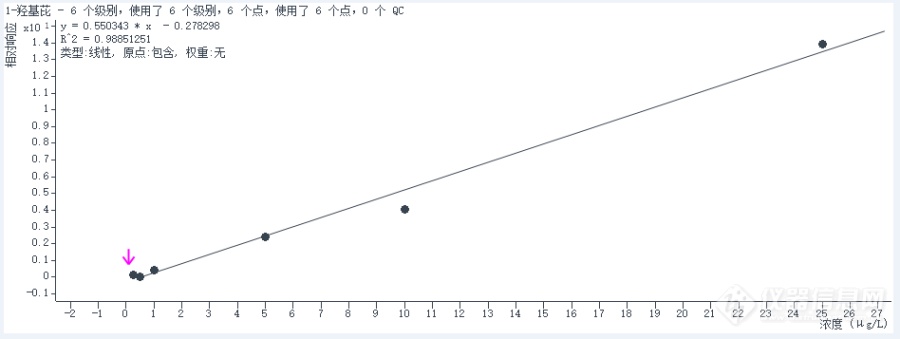
使用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱联用仪检测尿中多环芳烃代谢物,其中羟基代谢物1-羟基芘校正曲线的线性结果不好,R2=0.98,请问可能是什么原因引起的?[img=,690,259]https://ng1.17img.cn/bbsfiles/images/2023/08/202308030823508721_7973_5852764_3.png!w690x259.jpg[/img]
文献:气相色谱-质谱质谱法检测蔬菜中的毒死蜱及其代谢物
[color=#444444]最近用maldi-TOF MS做的数据,是分析铜绿微囊藻在不同处理条件下的代谢物质变化,了解到产脂肪酸、烃类、脂质类等类的物质,得到的质谱峰比较多,怎样确定这种藻产的代谢物质是什么呢[/color]
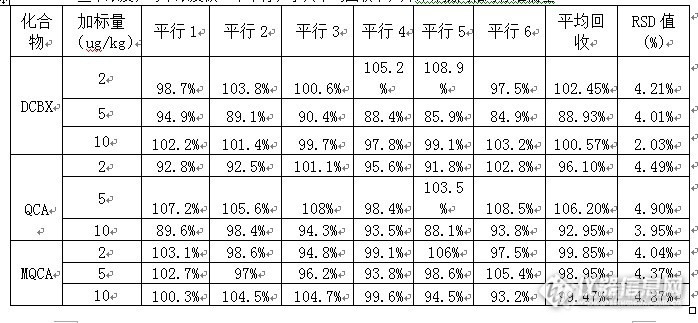
液相色谱串联质谱法测定动物组织中卡巴氧和喹乙醇代谢物残留量 摘要:采用高效液相色谱-电喷雾串联质谱仪(LC-ESI-MS-MS),建立了猪肉中卡巴氧代谢物脱氧卡巴氧、喹恶啉-2-羧酸和喹乙醇代谢物3-甲基喹恶啉-2-羧酸残留量的药物残留的检测方法,样品用甲酸溶液消化,蛋白酶水解,盐酸酸化,离心过滤后,过Oasis MAX固相萃取住或相当者净化。先用二氯甲烷洗脱脱氧卡巴氧,再用%甲酸乙酸乙酯溶液洗脱喹恶啉-2-羧酸和3-甲基喹恶啉-2-羧酸,氮气吹干洗脱液,残渣用甲酸+甲醇(19+1)溶液溶解,样液供液相色谱-串联质谱仪测定,内标法定量。本方法采用了2ug/kg,5ug/kg,10ug/kg,3个添加浓度,每个浓度6个平行样品,上述3种药物残留的回收率在80%~110%,相对偏差在2.03%~4.94%。关键词:液相色谱串联质谱法;脱氧卡巴氧;喹恶啉-2-羧酸;3-甲基喹恶啉-2-羧酸。1 引言卡巴氧(Carbadox) 和喹乙醇(Olaquindox) 同属喹喔啉类化合物 , 该类药物具有显著的促进动物生长的作用 , 用作猪等养殖动物的饲料添加剂。二者本身具有潜在的致畸变、致癌作用 , 其代谢物也可能带来健康风险。因此许多国家将以卡巴氧和喹乙醇列为对食用动物禁用或限用的药物 , 欧盟、中国、日本、美国、澳大利亚等对二者在动物组织内迅速代谢而产生的相应的代谢产物喹喔啉-2-羧酸(QCA)和 3-甲基喹喔啉-2-羧酸(MQ-CA)制定了残留监控的限量标准。在动物体内,喹乙醇和卡巴氧、经脱单氧、脱双氧后主要生成脱氧卡巴氧、喹恶啉-2-羧酸、3-甲基喹恶啉-2-羧酸,相对应的代谢物比较稳定,通常作为残留分析和监控的目标物质,代谢途径见图1。鉴于喹乙醇和卡巴氧的毒性和潜在的危害,为了更好的对动物食品进行监控,本文旨在建立喹乙醇、卡巴氧代谢物的残留液质联用仪检测方法。 http://ng1.17img.cn/bbsfiles/images/2013/10/201310242215_472741_2082444_3.jpg2 实验部分1.1仪器与试剂1.1.1试剂和材料甲醇:德国默克,色谱纯。乙腈:德国默克,色谱纯;乙酸乙酯:德国默克,色谱纯;水:1.25L哇哈哈纯净水(杭州产);正己烷:Honeywell,色谱纯。甲酸:色谱纯乙酸:色谱纯浓盐酸:分析纯乙酸钠:分析纯甲酸乙酸乙酯溶液:2% 向400mL乙酸乙酯中加入10mL甲酸,用乙酸乙酯定容至500mL。甲酸溶液
使用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱联用仪检测多环芳烃代谢物,其中1-羟基菲混标的校正曲线线性不好,R2只有0.98,请问是什么原因?有什么解决办法?
建立了牛奶中4 种安乃近代谢物—— 4- 甲酰氨基安替比林、4- 乙酰氨基安替比林、4- 氨基安替比林和4- 甲基氨基安替比林的液相色谱- 串连质谱(LC-M/MS)测定法。样品加入TRIS 缓冲溶液后用乙腈提取,提取液经乙腈饱和过的正己烷脱脂净化,采用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS 电喷雾正离子(ESI+)、多反应监测(MRM)模式检测,4- 乙酰氨基安替比林、4- 甲酰氨基安替比林和4- 氨基安替比林采用外标法定量,4- 甲基氨基安替比林采用内标法进行定量。4- 甲酰氨基安替比林的检出限为0.24μg/kg,4- 氨基安替比林的检出限为0.59μg/kg,4- 乙酰氨基安替比林的检出限为0.20μg/kg,4- 甲基氨基安替比林的检出限则为0.61μg/kg。在添加量5~20μg/kg 范围内,4 种安乃近的回收率在80.4%~97.9% 范围内,相对标准偏差(RSD)均小于9%。
新手,第一次做食品中硝基呋喃类药物的检测。请做过的老师们指点一下。用硝基呋喃类代谢物标准品优化质谱参数还需要衍生么,样品前处理衍生的目的是什么? 衍生的话,它们的母离子跟子离子质量数还跟国标上一致么?国标21311里面衍生,不同浓度的基质标曲加入的衍生剂的量相同。这样会不会影响衍生的效果?
http://img3.17img.cn/bbs/upfile/images/20100518/201005181701392921.gif质谱在环境污染物及其代谢物的检测中的应用讲座时间:2014年10月23日 14:00 主讲人:王海鉴AB SCIEX公司行业市场经理http://img3.17img.cn/bbs/upfile/images/20100518/201005181701392921.gif【简介】 质谱在环境污染物及其代谢物的检测中的应用,主要涉及药物,个人护理品 (PPCP)、全氟化合物(PFC)、丙烯酰胺类、多环芳烃 (PAH)、溴代阻燃剂 (BFR)、环境毒素(如,微囊藻毒素等)、消毒副产物等。 解决方案有多组分同时检测、使用离子丰度比例或者二级谱库比对定性、使用高分辨质谱进行未知物筛查等。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2014年10月23日 13:304、报名参会:http://simg.instrument.com.cn/meeting/images/20100414/baoming.jpg
中药代谢物鉴定采用直观比较判别法、MDF MetaboLynx数据处理技术、主成分分析法、离子淌度质谱分析法,到底那种技术好?
如题,请问我这边买的标准品已经是代谢物了,还需要经过衍生化后才能上质谱优化吗?还是可以不衍生直接用质谱优化?
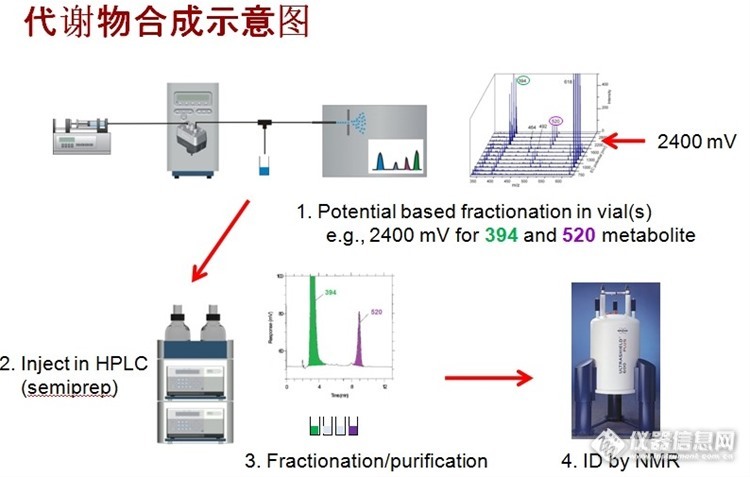
在新药筛选时,代谢物往往没有标准品,要得到足够量的代谢物进行结构认定等研究也比较困难,ROXY电化学可解决这个难题,可快速制备代谢物(降解产物)用于质谱,核磁分析或作为标准参考物。 用3 - 甲氧基-4 - hydroxyphenylglycol(MOPEG)证明了此原理。在10分钟内,0.1 mmol / L的MOPEG(1.4毫克)几乎完全转化。http://ng1.17img.cn/bbsfiles/images/2014/11/201411051614_522048_1617240_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411051614_522049_1617240_3.jpg
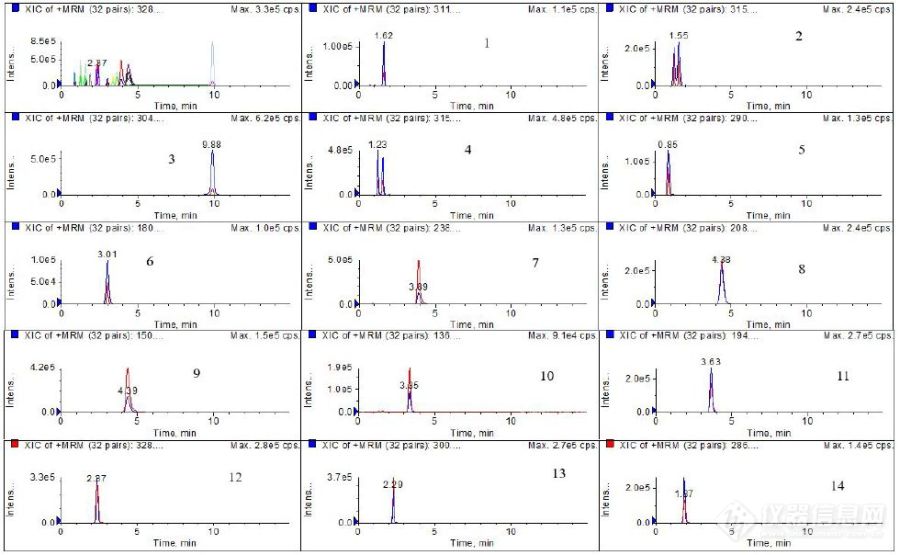
[align=left]该方案按照《司法鉴定技术规范 SF/Z JD0107025-2018 毛发中15种毒品及其代谢物的液相色谱-串联质谱检验方法》,分别使用CAPCELL PAK PFP和适合质谱分析的CAPCELL PAK C18 MGIII对除“去甲氯胺酮”之外的14种毒品混标溶液进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]分析,两种色谱柱均可实现14种毒品的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]良好分析。[/align][align=left][color=black]CAPCELLPAK PFP[/color][color=black]色谱柱键合五氟苯基官能团,采用比表面积较大的全多孔型硅胶作为基材,具有更好的分离能力;提高了官能团的键合密度,能与分析对象间产生较强的相互作用,因此在异构体的拆分中能展现出优越的识别能力。[/color][/align][align=left][color=black]CAPCELLPAK C18 MGIII[/color][color=black]色谱柱使用高纯度硅胶基材,通过减少硅胶微细孔的数量来增大有效比表面积,从而达到良好的分离效果。着眼于“在酸性条件下对碱性化合物的高重现性稳定分析”,对填料进行了特殊的预处理,背景噪音极低、柱流失极低,适用于[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]分析。[/color][/align][color=black][/color][align=left][/align][color=black][/color][align=left][/align][align=left][b][color=#0070c0]方法一CAPCELL PAK PFP分析14种毒品及代谢物[/color][/b][/align][align=left]该方法按照《司法鉴定技术规范 SF/Z JD0107025-2018 毛发中15种毒品及其代谢物的液相色谱-串联质谱检验方法》,使用CAPCELL PAK PFP色谱柱,调整流速(由350μl/min调整为500μl/min,压力不超过15Mpa)后,可实现14种毒品(除去“去甲氯胺酮”)的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]良好分析。[/align][align=left][img=,400,248]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281454435401_3453_2222981_3.jpg!w629x390.jpg[/img] [img=,400,246]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281455283985_5910_2222981_3.jpg!w900x555.jpg[/img][/align][align=left][img=,600,142]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281455526505_1029_2222981_3.jpg!w900x213.jpg[/img][/align][align=left]CAPCELL PAK PFP分析14种毒品的MRM色谱图(500 μL/min)[/align][align=left]1. 大麻酚 2. 大麻二酚 3. 可卡因 4. △[sup]9[/sup]-四氢大麻酚 5. 苯甲酰爱康宁 6. MDA 7. 氯胺酮 8. MDEA 9. MAMP 10. AMP 11. MDMA 12. O[sup]6[/sup]-单乙酰吗啡 13. 可待因 14. 吗啡[/align][align=left][b][color=#0070c0] [/color][/b][/align][align=left][b][color=#0070c0]方法二CAPCELL PAK C18 MGIII分析14种毒品及代谢物[/color][/b][/align][align=left]该方法基于《司法鉴定技术规范 SF/Z JD0107025-2018 毛发中15种毒品及其代谢物的液相色谱-串联质谱检验方法》,使用适合质谱分析的CAPCELL PAK C18 MGIII,调整梯度洗脱程序,可实现14种毒品(除去“去甲氯胺酮”)的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]良好分析。[/align][align=left][img=,400,247]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281456127401_1959_2222981_3.jpg!w631x391.jpg[/img] [img=,400,247]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281456297935_1710_2222981_3.jpg!w900x557.jpg[/img][/align][align=left][img=,600,143]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281456460031_9213_2222981_3.jpg!w900x215.jpg[/img][/align][align=left]CAPCELL PAK C18 MGIII分析14种毒品的MRM色谱图(350μL/min)[/align][align=left]1. MAMP 2. 苯甲酰爱康宁 3. 大麻二酚 4. △[sup]9[/sup]-四氢大麻酚 5.大麻酚 6. MDA 7. 可卡因8. MDEA 9. 可待因 10. 吗啡 11. 氯胺酮 12. O[sup]6[/sup]-单乙酰吗啡 13. MDMA 14. AMP[/align][align=left][color=black]综上实验结果,使用五氟苯基色谱柱CAPCELL PAK PFP和适用于质谱分析的CAPCELL PAK C18 MGIII,基于司法鉴定技术规范方法,均可实现14种毒品及代谢物的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]良好分析。[/color][/align][align=left][color=black]附表1 14种毒品样品的定性离子对及保留时间[/color][/align][align=left][color=black][img=,500,682]https://ng1.17img.cn/bbsfiles/images/2019/03/201903281457250471_7884_2222981_3.jpg!w900x1229.jpg[/img][/color][/align][align=left][/align][align=left][/align]
如题,想找几篇文献,找找方法,如果您分析TCA循环中的代谢物,请帮个忙,谢谢
我实验室质谱是LTQ orbitrap, 主要用来做蛋白组学,最近有客户要做氨基酸代谢物定量分析,我建立分析方法,采用一级MS全扫,提取离子定量,做标准曲线(不含基质)发现6个物质只有一个线性在99%以上,其余在96-99之间,标曲6个点最高2ug/ml,最低2ng/ml, 在高浓度区域的点(2ug,1ug)总是在线性下面,响应偏差,形成往上抛物线状,高中低浓度的精密度均很好。是不是浓度过高造成离子抑制,做了些改进,在样品和流动相中均提高甲酸浓度,并在离子源部分提高气流,电压等,线性并未改善。我的样品采用5%DMSO溶解(含甲酸, 不好溶解,所以用DMSO), 流动相采取乙腈-水(含甲酸)。我看到很多文章在用此方法标曲线性均在99以上,可是我总是达不到。是不是离子阱不适合分析这类样品,或者确实是非线性,要用加权最小二乘进行处理。各位有什么建议,做哪些改进能提高线性呢?先行谢过。
随着生化学科的发展,肽类药物必然要越来越受到大家的重视,但肽类的质谱分析却是比较难的,大家都来谈谈肽类药物质谱分析的经验。我有个同学在坐一个肽类化合物,代谢物从分析液相接出来,质谱直接进样,却没有响应,大家说如何提高它的响应呢?
各位老师,大神好,最近我有在做农作物的代谢组学检测,参考一篇论文的方法进行的,他把代谢物分为两大部分来做,然后当我在做极性物检测的时候,对氨基酸和有机酸的检测经常出现问题,除了一种苹果酸外,其他检测不到。希望有人做代谢物的进行一个交流,谢谢
1.前言 随着科学与技术的发展,新药研发的速度正在日益加快,使得新药安全性评价工作的压力也变得越来越大。在新药研究开发过程中,因为安全性问题而被淘汰的候选药物占相当大的比例。一旦潜在的药物分子通过了初步的生物学筛选过程,就应该尽量减少这些候选药物分子在产品研发过程中的流失,以免造成巨大的资金和时间的浪费。因此,人们努力寻找新的分析方法,以便从功效和安全性两方面使得先导化合物的筛选更有效,从而尽可能地减少这种浪费。目前的生物分析手段主要利用基因组和蛋白质组方法,分别从基因水平和细胞蛋白质表达水平上测量生物体系对药物的反应。这两种方法都较昂贵,且劳动强度较大,然而却可能是研究在不同水平上对生物异源物质的生物应答的有力工具。但是,基因组学和蛋白质组学都不能提供可以了解生物体中整体细胞功能的信息,因为两者都忽略了整体器官中动态的代谢状态。因此,Nicholson等人提出了一种基于核磁共振的新方法,叫做metabonomics,我们暂且称之为代谢组学,以便与由代谢物组(metabolome)衍生而来的metabolomics相区别。Metabolomics研究的是一个细胞或细胞类型中所有的小分子成分,而metabonomics则是通过分析生物体液和组织来对完整的生物体(而不是单个细胞)中随时间改变的代谢物进行检测、确定、定量和分类;然后将这些***代谢轨迹与病生理过程中的生物学事件关联起。从药物研究和毒理学评价的角度来看,基因组学方法是观察给药后基因表达的改变,主要采用基因芯片技术。然而,基因调节/表达与系统的整体功能之间的关系在目前还很不清楚,主要是因为决大部分DNA是非编码的,而编码蛋白质的基因不能孤立地发挥作用,而是需要与其邻近的基因和非编码DNA一起才能发挥其功能。正式由于这个原因,人们才发展了蛋白质组学。蛋白质组学方法可以对由给药或其它病生理过程引起的细胞蛋白质组成变化进行半定量的测量。蛋白质组方法所采用的技术主要包括双向凝胶电泳和质谱技术。与基因组方法相比,蛋白质组方法较慢,且劳动强度较大。需要强调的是,虽然这些方法能够在很大程度上揭示毒理学机理,并且给出与疾病相关的新的生物标记物,却很难将这些发现与经典的毒理学指标相关联。原因很简单,因为目前的技术和方法不能对给药后反应的整个进程进行测量,也不能对生物整体的应答进行测量。因此需要发展一种新的方法来实时给出多器官生物整体的在体信息。基于NMR的代谢组学(metabonomics)方法可以满足这样的要求。2.Metabonomics在药物毒理学研究中的应用 代谢组学的目的是要扩展和补充由基因组学和蛋白质组学方法得到的对生物异源物质应答的信息。其任务是定量测量生物体对病生理刺激或基因改变的动态多参数代谢反应,是研究药物毒性和基因功能的技术平台。这个概念是根据Nicholson小组近二十年来利用1H NMR技术研究生物体液、细胞和组织中多组分代谢组成的工作而提出的。在这些研究中,还利用了模式识别,专家系统和相关的生物信息学工具。在许多情况下,药物通过与遗传物质直接作用而产生毒性,或通过诱导系统合成与药物代谢有关的酶,从而产生有毒的产物。在这种情况下,用基因组和蛋白质组学方法来评价毒性是有用的。然而,在生物异源物质有可能只在药理学水平上产生作用,因而可能不会影响基因的调节和表达。再者,显著的毒理学效应可能与基因的改变和蛋白质的合成完全不相关。因此,在许多情况下,从基因组和蛋白质组角度考虑到的反应可能不能预测药物毒性。但是,所有的由药物引起的病生理紊乱都会由于直接的化学反应,或通过与控制代谢的酶或核酸相结合而引起内源生化物质在比例、浓度、代谢通量等方面的失调。如果这种变化足够大的话,就会影响整个生物体的功能。生物体液中的代谢物是与细胞和组织中的代谢物处于动态平衡,因此,生物体中由于中毒或代谢损害而引起的细胞功能异常一定会反映在生物体液成分的变化中。要检测血浆、尿液、胆汁等生物基质中的一些具有特殊意义的微量物质,选择合适的分析方法致关重要。高分辨1H NMR波谱就非常适合用来检测生物体液中的成分异常,因为该方法可以同时对所有的代谢物进行定量分析,而且不需要样品前期准备,对任何成分一样灵敏。虽然也可以采用如质谱等其它方法,但对不同成分离子化程度的差别会影响定量和检测的可靠性。NMR方法还可以有效地用来从组织萃取物或细胞悬液中找出异常的代谢物。还可以利用高分辨魔角旋转(HR-MAS)探头来检测完整组织中的代谢物组成。由1H NMR谱检测到的生物体液中的内源性代谢物模式完全依赖于动物体内的毒素的类型。每一种类型的毒物都会在生物体液中产生特征的内源代谢物浓度和模式变化,这种特征给我们提供了毒性作用的机理和毒性位置的信息。右图所示为一系列尿样的1H NMR谱图,是大鼠经不同的毒物处理后得到的。每一张谱图只需几分钟的时间,是非常有效的。可以看出,不同毒素引起的代谢物变化是有特征性的。因为几乎所有的代谢物都有其特征的NMR谱,因而可以作为毒物引起的代谢变化的指纹图谱。利用NMR方法,人们已经成功地发现了许多新的器官特异相关毒性的代谢标记物。作为分析生物化学技术,NMR正是在这种探索性的工作上具有优势。
摘要:建立了可以同时检测动物源性食品中喹乙醇代谢物—3-甲基喹噁啉-2-羧酸,和卡巴氧代谢物—喹噁啉-2-羧酸的固相萃取-气相色谱质谱联用检测法。样品经偏磷酸甲醇水解,乙酸乙酯萃取和磷酸盐缓冲液反萃取,用混合阴离子交换SPE柱净化后,洗脱液氮气吹干, N,N-二甲基甲酰胺二甲基缩醛衍生,衍生产物直接进气相色谱质谱联用仪检测。通过对猪肉、鸡肉和鸡蛋的添加回收试验,考察了方法的准确度、精密度和基质适应性。结果显示:目标分析物分别在0.5~5μg/kg质量浓度范围内呈良好线性,相关系数不低于0.9985;方法回收率达到87%~107%,相对标准偏差均小于15%;3-甲基喹噁啉-2-羧酸的检出限达到0.5μg/kg,甲基喹噁啉-2-羧酸的检出限达到0.2ug/kg;方法基质适应性良好。关键词:喹乙醇代谢物;3-甲基喹噁啉-2-羧酸;卡巴氧代谢物;喹噁啉-2-羧酸;甲酯化衍生;固相萃取;气相色谱质谱联用喹噁啉类兽药包括卡巴氧、喹乙醇、喹烯酮等品种。卡巴氧、喹乙醇曾作为抗菌促生长剂在养殖业中都得到广泛的应用。根据JECFA报告中对卡巴氧和喹乙醇的风险评估,不仅二者本身具有潜在的致畸变、致癌作用,其代谢物也可能带来健康风险。因此许多国家将卡巴氧和喹乙醇列为对食用动物禁用或限用的药物,毒理实验研究表明,喹噁啉类兽药具光敏毒性、致畸性和致癌性,欧盟已于1998年禁止其应用于可食性动物,中国兽药典(2005)明确规定,禁止在家禽及水产养殖中使用喹乙醇,但仍允许使用喹乙醇作为猪饲料添加剂。在动物体内,卡巴氧和喹乙醇经脱单氧、脱双氧后主要生成喹噁啉2-羧酸(QCA)或3-甲基喹噁啉2-羧酸(MQCA)。由于二者的原药在动物组织内代谢迅速(代谢途径见图1),而相应的代谢产物喹噁啉-2-羧酸(QCA)和3-甲基喹噁啉-2-羧酸(MQCA)则相对稳定,因此通常将这两个代谢产物作为残留分析和监控的目标物。1995年联合国粮农组织(FAO)/世界卫生组织(WHO)食品添加剂联合专家委员会(JECFA)将3-甲基喹噁啉2-羧酸(MQCA)规定为喹乙醇在动物组织中的残留标识物。2003年我国农业部规定了肌肉和肝脏组织中MQCA的最大残留限量分别为[fon
[align=center][size=16px]人血清中[/size][size=16px]多[/size][size=16px]种农兽药及其代谢物的高分辨、高通量检测技术与方法学研究[/size][/align][size=16px]通过制备具有[/size][size=16px]多功能化超大比表面积[/size][size=16px]的[/size][size=16px]CMPs[/size][size=16px]和桥连硅烷化试剂修饰磁性纳米富集净化材料,并将其制备成萃取分离装置,构建集样品富集浓缩、高效净化的集成化前处理平台。进一步利用[/size][size=16px]LC-QTOF-MS[/size][size=16px]和[/size][size=16px]LC-Q Exactive Orbitrap-MS/MS[/size][size=16px]高分辨质谱,建立[/size][size=16px]300[/size][size=16px]种以上农兽药及其主要代谢物的标准质谱匹配信息库和保留时间数据库,并初步用于京津冀地区人血清样本中农兽药及代谢物的全面筛查,构建不同检测技术下农兽药化学污染物在人体内蓄积水平数据库,实现农兽药残留数据获取、污染等级判定、多维度表达及分析的自动化,为后续规模化样品分析和农兽药残留与疾病的关联性分析提供技术支撑。[/size][size=16px]2.1 [/size][size=16px]研究内容[/size][size=16px]([/size][size=16px]1[/size][size=16px])人血清中农兽药及代谢物高效净化技术研究[/size][size=16px]针对[/size][size=16px]人血清中农兽药及代谢物[/size][size=16px]残留水平低、结构特性多样等特征[/size][size=16px],以多粒径磁性纳米粒子为基质,制备具有超大比表面积的[/size][size=16px]桥连硅烷化试剂修饰材料和[/size][size=16px]共轭微孔聚合物材料,提高农兽药及[/size][size=16px]其代谢物的富集效率;通过修饰引入多羟基、磺酸基、卤代烃等多功能化基团,实现[/size][size=16px]不同结构特征[/size][size=16px]农兽药及其代谢物的高通量富集;将研制的富集净化材料进一步制备成萃取分离装置,构建集样品富集浓缩、高效净化的集成化前处理平台,实现人血清中农兽药及代谢物的高通量靶向[/size][size=16px]/[/size][size=16px]非靶向富集净化。[/size][size=16px]([/size][size=16px]2[/size][size=16px])人[/size][size=16px]血清中农兽药及代谢物的高通量筛查与定量分析方法研究[/size][size=16px]针对人血清中农兽药代谢复杂、残留水平较低的特点,利用[/size][size=16px]LC-QTOF-MS[/size][size=16px]和[/size][size=16px]LC-Q Exactive Orbitrap-MS/MS[/size][size=16px]高分辨质谱,建立[/size][size=16px]300[/size][size=16px]种以上农兽药及主要代谢物的标准质谱匹配信息库和保留时间数据库;结合课题开发的高通量农兽药残留分离富集前处理技术,建立覆盖[/size][size=16px]300[/size][size=16px]种以上农兽药及代谢物的血清样品高通量非靶向高分辨质谱检测技术,实现人血清农兽药残留由靶向检测向非靶向筛查的跨跃式发展;对所建立的方法进行方法学验证评价,形成方法标准操作规程。[/size][size=16px]([/size][size=16px]3[/size][size=16px])人血清样本中农兽药及代谢物全面筛查与数据库构建[/size][size=16px]基于所研发的人血清中[/size][size=16px]300[/size][size=16px]种农兽药及代谢物高通量非靶向高分辨质谱检测技术,开展京津冀地区人血清样本中农兽药及代谢物的初步筛查,构建不同检测技术下农兽药化学污染物在人体内蓄积的数据库,实现农兽药残留数据获取、污染等级判定、多维度表达及分析的自动化[/size][size=16px],[/size][size=16px]为后续规模化样品分析和农兽药残留与疾病的关联性分析提供技术支撑。[/size][size=16px]2.2 [/size][size=16px]研究目标[/size][size=16px]([/size][size=16px]1[/size][size=16px])针对人血清基质复杂,农兽药及代谢物残留浓度低、形态多样化等特点,筛选并制备出[/size][size=16px]多功能化超大比表面积磁性纳米材料[/size][size=16px],[/size][size=16px]实现不同结构特征的农兽药及其代谢物的[/size][size=16px]高效、[/size][size=16px]高通量富集[/size][size=16px]。[/size][size=16px]([/size][size=16px]2[/size][size=16px])[/size][size=16px]建立人血清中农兽药及其代谢物的高通量非靶向高分辨质谱检测技术,实现农兽药及其代谢物的快速筛查与精准定量分析。[/size][size=16px]([/size][size=16px]3[/size][size=16px])通过结合开发的高通量非靶向检测技术,初步用于京津冀地区人血清[/size][size=16px]中[/size][size=16px]农兽药的高通量非靶向高分辨质谱检测,构建人血清中农兽药残留蓄积形态数据库,筛查出高残留的农兽药及其代谢物。[/size][size=16px]([/size][size=16px]1[/size][size=16px])[/size][size=16px]通过设计开发[/size][size=16px]多功能化超大比表面积磁性纳米材料[/size][size=16px],实现[/size][size=16px]复杂[/size][size=16px]的人[/size][size=16px]血清[/size][size=16px]样本中[/size][size=16px]结构特征[/size][size=16px]多样[/size][size=16px]的农兽药及其代谢物[/size][size=16px]高效富集和净化。[/size][size=16px]([/size][size=16px]2[/size][size=16px])针对农兽药及代谢物种类繁多、性质各异限制其高通量非靶向筛查的问题,通过建立[/size][size=16px]300[/size][size=16px]种以上农兽药及代谢物的标准质谱匹配信息库和保留时间数据库,为农兽药高通量非靶向筛查方法的研发奠定基础。[/size]
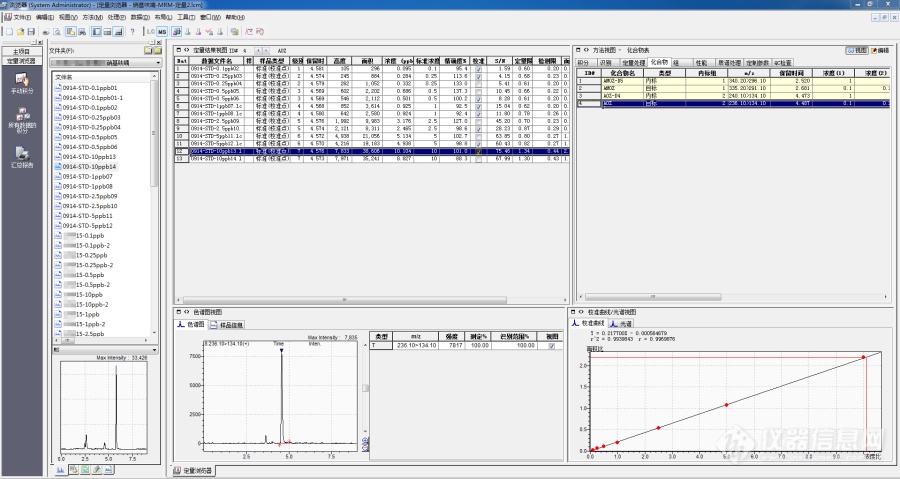
[b][b]液相色谱-串联质谱法测定草鱼中硝基呋喃类代谢物AOZ残留量[/b]摘要[/b]: 本文根据农业部783号公告-1-2006水产品中硝基呋喃类代谢物残留量的测定 液相色谱-串联质谱法来检测草鱼中硝基呋喃类代谢物AOZ的残留。样品经水解、衍生化和净化后,采用多反应监测模式测定。通过化合物的保留时间、定性离子和定量离子的筛查与确证,实现对草鱼中硝基呋喃类代谢物AOZ的定性与定量。结果表明,其线性系数大于0.99,在2.5mg/kg和4.5mg/kg添加水平下,AOZ回收率在95%-100%之间,相对标准偏差(RSD)小于15%。样品经水解、衍生化和净化后,以流动相A:0.002mol/L的醋酸铵溶液,B:甲醇进行梯度洗脱,C18(100mm×2.1mm,i.d,5um)色谱柱分离,采用正离子多反应监测( MRM) 模式检测,基质匹配标准溶液定量。线性范围内相关系数均大于[color=#ff0000] [/color]0. 99。加标回收率在95.00%-100% 之间,相对标准偏差在6.19%- 8.43% 之间,结果表明,该方法简便、快速、灵敏度高、重现性好,可测定草鱼中硝基呋喃类代谢物AOZ的残留。[b]关键词[/b]: 高效液相色谱 - 串联质谱 硝基呋喃代谢物AOZ 草鱼;[b]Abstract[/b]:In this paper, AOZ residue of nitrofuran metabolites in grass carp was detected by liquid chromatography-tandem mass spectrometry according to the determination of nitrofuran metabolites residues in aquatic products in the ministry of agriculture no.783 bulletin 1-2006. After hydrolysis, derivatization and purification, the samples were determined by multi-reaction monitoring. AOZ, a nitrofuran metabolite in grass carp, was qualitatively and quantitatively determined by retention time, qualitative and quantitative ion screening. The results showed that the linear coefficient was greater than 0.99, the recovery of AOZ was between 95% and 100% and the relative standard deviation (RSD) was less than 15% at the addition levels of 2.5mg/kg and 4.5mg/kg.After the samples were hydrolyzed, derivated and purified, the samples were separated by gradient elution in mobile phase A:0.002mol/L ammonium acetate solution,B: methanol, and separated by C18(100mm×2.1mm, i.d,5um) chromatographic column. Positive ion multireaction monitoring (MRM) mode was adopted for detection, and the matrix matched standard solution quantification. The correlation coefficients in the linear range are all greater than 0.99. The standard recovery was between 95.00% and 100%, and the relative standard deviation was between 6.19% and 8.43%.[b]Key words[/b]:ultra performance liquid chromatography-tandem mass spectrometry;nitrofuran metabolites-AOZ Grass carp.[b]1 实验部分[/b]1.1 仪器与试剂液相色谱—质谱联用仪( 岛津[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]-8030) 超纯水系统(WP-UP-YJ-20, 沃特浦) 分析天平(Quintix 2102-1CN,赛多利斯公司);旋涡混合器(VORTEX 3,IKA公司);恒温水浴振荡器(江苏安普SHA-C);离心机(湖南湘仪H-2050R);甲醇(色谱纯,默克股份两合公司) 醋酸铵( 分析纯,天津市致远化学试剂有限公司);2-硝基苯甲醛(色谱纯,天津市百世化工有限公司);二甲基亚砜(分析纯,天津市百世化工有限公司);磷酸氢二钾(分析纯,天津市百世化工有限公司);乙酸乙酯(分析纯,天津市泰兴试剂厂);农药标准物质: 均来自农业部环境保护科研监测所(天津);实验用水为经沃特浦超纯水系统处理后的超纯水。1.2 [b]样品处理[/b]1.2.1[b] 样品水解与衍生化[/b] 准确称取样品2.0g(精确至 0.01 g) 到50 mL 离心管中,加入0.05mL的100ng/mL混合内标(AOZ-D[sub]4[/sub]),旋涡混合50s,再加入5mL盐酸溶液(0.2mol/L)和0.15mL的2-硝基苯甲醛溶液(0.05mol/L),涡旋振荡50s后,置于恒温水浴振荡器中37℃避光震荡16h。1.2.2 [b]样品的提取净化[/b]取出离心管冷却至室温,加入3-5mL磷酸氢二钾溶液(1.0mol/L),调节pH至7.0-7.5,加入4mL乙酸乙酯,涡旋振荡50s,4000r/min离心5min,取上层清液转移至10mL玻璃离心管中;再加入4mL乙酸乙酯重复上述操作,合并上清液于40℃下氮气吹干。加入1.0mL甲醇溶液(甲醇:水=5:95(V:V))涡旋振荡溶解残留物,过0.45um滤膜,待测。1.2.3 [b]溶液配制 [/b](1)[b]标准储备液。[/b] 分别取上述标准品(100.0 ug/mL 溶液),用甲醇稀释成 10 ng/mL 和100 ng/mL的标准储备液。(2)[b]标准工作溶液。[/b] 分别吸取上述标准品10ng/mL的标准储备液0.010mL、0.025mL、0.050mL、0.10mL和100ng/mL的标准储备液0.025mL、0.05mL、0.10mL于7个50mL离心管中,不加样品,按照1.2.1和1.2.2步骤操作,按照1.3测定。1.3 [b]色谱 - 质谱条件[/b]1.3.1 [b]色谱条件 [/b] 色谱柱:C18柱,(100mm × 2.1mm,5μm ) 柱温为40℃;进样量为20μL。流动相:A.0.002mol/L的醋酸铵溶液,B:甲醇 色谱洗脱条件见表 1。[align=center]表 1 高效液相色谱梯度洗脱条件[/align][table][tr][td][align=center]时间,min[/align][/td][td][align=center]A,%[/align][/td][td][align=center]B,%[/align][/td][td][align=center]流速,mL. [/align][/td][/tr][tr][td][align=center]3.5[/align][/td][td][align=center]15[/align][/td][td][align=center]85[/align][/td][td][align=center]0.25[/align][/td][/tr][tr][td][align=center]5[/align][/td][td][align=center]15[/align][/td][td][align=center]85[/align][/td][td][align=center]0.25[/align][/td][/tr][tr][td][align=center]5.1[/align][/td][td][align=center]76[/align][/td][td][align=center]24[/align][/td][td][align=center]0.25[/align][/td][/tr][/table]1.3.2[b]质谱条件[/b]离子源: 电喷雾离子源ESI 扫描模式为正离子扫描 雾化气流量3L/min;干燥气流量15L/min;加热块温度250℃;DL温度250℃;CID气230kPa;IG真空度1.9×10[sup]-3[/sup]pa;PG真空度7.5×10[sup]1[/sup]pa;检测方式为多重反应监测。其他质谱参数见表2。 [align=center]表 2 反应监测的质谱采集参数[/align][table][tr][td][align=center]化合物[/align][/td][td][align=center]母离子m/z[/align][/td][td][align=center]子离子m/z[/align][/td][td][align=center]碰撞能量(v)[/align][/td][/tr][tr][td=1,2][align=center]AOZ[/align][/td][td][align=center]236[/align][/td][td][align=center]104[/align][/td][td][align=center]19[/align][/td][/tr][tr][td][align=center]236[/align][/td][td][align=center]134*[/align][/td][td][align=center]22[/align][/td][/tr][tr][td][align=center]AOZ-[/align][/td][td][align=center]240[/align][/td][td][align=center]134*[/align][/td][td][align=center]14[/align][/td][/tr][tr][td=4,1]注:*为定量碎片离子[/td][/tr][/table][b]1.4 添加回收实验[/b] 准确称取不含上述药物的草鱼样品2.0g,分别以2.5mg/kg和4.5mg/kg2个水平进行添加回收实验,重复5次,按照上述实验方法测定,计算添加回收率和相对标准偏差。[b]2 结果与讨论2.1线性回归方程[/b]AOZ的基质标准曲线为 y = 0.217700 x -0.000564679( r = 0.9969876,r[sup]2[/sup]=0.9939843) 如图3 [align=center]图3(标准曲线AOZ)[/align][img=图片3 标准曲线,690,367]https://ng1.17img.cn/bbsfiles/images/2019/10/201910100829478357_6345_3416090_3.png!w690x367.jpg[/img][b]2.2方法的准确度、精密度[/b]在空白草鱼中分别进行 2.5和4.5 mg / kg 2 个水平的加标回收试验,每个水平重复测定 5 次,计算加标回收率和相对标准偏差。由表 5 可知,在 2.5和4.5mg / kg 2个添加水平下,AOZ的平均回收率分别为 99.648% 和95.63% ,相对标准偏差分别为 8.43% 和6.19%,方法显示出良好的准确度和精密度,可以满足试剂样品中农药残留的检测要求。[align=center]表 5 AOZ在草鱼中的回收率和相对标准偏差[/align][table][tr][td][align=center]农药[/align][/td][td][align=center]添加浓度( mg / kg)[/align][/td][td][align=center]平均回收率( %)[/align][/td][td][align=center]重复 1[/align][/td][td][align=center]重复 2[/align][/td][td][align=center]重复 3[/align][/td][td][align=center]重复 4[/align][/td][td][align=center]重复 5[/align][/td][td][align=center]平均值相对标准偏差( % )[/align][/td][td][align=center]标准曲线[/align][/td][td][align=center]相关系数r2[/align][/td][/tr][tr][td]AOZ[/td][td]2.5[/td][td]99.648[/td][td]2.517[/td][td]2.261[/td][td]2.663[/td][td]2.723[/td][td]2.292[/td][td]8.43[/td][td][align=center]Y=0.217700X-0.000564679[/align][/td][td]0.9939843[/td][/tr][tr][td]AOZ[/td][td]4.5[/td][td]95.63[/td][td]4.223[/td][td]4.768[/td][td]4.043[/td][td]3.870[/td][td]4.613[/td][td]6.19[/td][td][align=center]Y=0.217700X-0.000564679[/align][/td][td]0.9939843[/td][/tr][/table][b]2.3实际样品分析[/b] 用本方法对市场销售的2份草鱼进行硝基呋喃类代谢物AOZ检测,均未检出,见图6。 [align=center]图6[/align][img=图片6,690,366]https://ng1.17img.cn/bbsfiles/images/2019/10/201910100830145934_7979_3416090_3.png!w690x366.jpg[/img][b]3、结论[/b]本研究通过对样品前处理方法、质谱条件和色谱条件的优化,建立了液相色谱-串联质谱法测定草鱼中AOZ残留量的分析方法,该方法前处理简单、快速、回收率高,方法的灵敏度、准确度和精密度等均满足农药残留分析的要求,适用于大量样品的快速检测。[b]参考文献[/b]高洁,朱莉萍等.超高效液相色谱-串联质谱法测定多脂肪类动物源性食品中硝基呋喃代谢物. 食品安全质量检测学报2018年第9卷第6期,2018.[b] [/b]
[b]【序号】:6【作者】:【题名】:[b][b][size=12px][font=&][/font][/size][font=Verdana, Arial][size=20px][color=#333333]DB22/T 1614-2012 蛋及乳制品中硝基呋喃代谢物残留量的测定 [url=https://insevent.instrument.com.cn/t/5p]液相色谱[/url]-质谱/质谱法[/color][/size][/font][font=&][color=#333333][/color][/font][/b][/b]【期刊】:【年、卷、期、起止页码】:【全文链接】:[/b]
最近在做硝基呋喃代谢物,因为刚开始接触质谱,对阳性样品判断不能肯定!想请教高手指点怎么判断!
目的使用ACQUITY UPLC®/Xevo™ G2 QTof质谱系统及MetaboLynx™ XS应用管理软件,鉴定通过人肝微粒体体外孵育而获取的1 μM维拉帕米的代谢物。背景近年来,随着越来越多的一线药品因存在安全性顾虑而退出市场,人们对药品研发过程中的药物代谢和毒性研究给予了更多的关注。如今,在药物发现和研制阶段提早进行药物代谢研究的趋势已比较明显。普遍的做法是对母体药物进行体外代谢物研究,以便在药品开发早期迅速确定其弱点。在药物发现阶段进行代谢物鉴定的一项挑战是:需要提供快速而通用的方法,并且该方法应足够灵敏,以使体外孵育研究可在低μM浓度水平下进行,从而使其更接近于化合物的体内作用情况。一项典型的体外代谢研究还包括分析母体药物的代谢速率和途径。此类研究的理想分析方案需提供在模拟体内条件的底物浓度下对代谢物进行检测的分析速度和灵敏度。利用与UPLC/MSE联用的Xevo G2 QTof质谱系统,体外代谢物研究可在低μM水平下进行,同时具有较好的速度、灵敏度和选择性。http://www.bio-equip.com/imgatl/20115514946.jpg图1. 人肝微粒体维拉帕米(1μM)的孵育结果显示在MetaboLynx浏览器中。解决方案将浓度为1 μM的维拉帕米与人肝微粒体在37°C下进行孵育,并分别在 0、15、30、60、120和 240分钟时加入等体积的冷乙腈终止反应。对样品进行离心,并取上清液直接进样。采用沃特世ACQUITY UPLC®系统,ACQUITY UPLC HSS T3色谱柱(1.7 μm、2.1 x 100 mm),进行色谱分离。流动相由0.1%甲酸水溶液(A)和乙腈(B)组成,进样量为5.0 μL。在ESI正离子模式下,使用Xevo G2 QTof质谱仪采用UPLC/MSE技术进行数据采集,这样一次进样即可同时获取母离子和产物离子的数据。MetaboLynx XS应用管理软件用于进行数据挖掘,结果显示在MetaboLynx浏览器中(如图1所示)。产物离子信息同时进行处理,并显示在MetaboLynx浏览器中的碎片分析窗口内(图2)。通过对多个孵育时间点的样品进样分析,母体药物的清除曲线和代谢物的形成曲线可在同一次试验中同时获取(如图3所示)。http://www.bio-equip.com/imgatl/20115514643.jpg图2. 碎片分析窗口中所显示的MS/MS信息。http://www.bio-equip.com/imgatl/20115514816.jpg图3. 维拉帕米的清除曲线(3A)及其代谢物的形成曲线(3B)。通过采用UPLC/MSE 数据采集策略,再加上具有化学智能的MetaboLynx XS数据处理工作流程,只需进行一次液相色谱进样即可快速完成所有代谢物的鉴定工作。通过在多个时间点进样,可比较容易地获取低浓度(μM)孵育水平下目标药物的代谢速率和途径。因此,产能最大化的目标即可轻松实现。总结这个应用表明:通过使用配备UPLC/MSE 和MetaboLynx XS工作流程的Xevo G2 QTof质谱系统,体外代谢物研究可在低浓度(μM)水平下进行,同时具有较好的速度、灵敏度和选择性。
本人新手,准备做呋喃唑酮代谢物AOZ的检测,用LC-MS做SRM。现在对于优化质谱条件有些不解,优化的时候优化的是AOZ的条件,还是AOZ衍生之后的条件?如果是衍生之后的话,我手里只有AOZ的标准品,应该怎样操作呢?第一次接触衍生,希望大家帮帮我。
请教大家一个问题,我们做硝基呋喃代谢物的检测时,四种物质只有AMOZ的线性可以,回收也可以,其他的三个线性不好,回收也不好。但是仪器上质谱图出的都挺好的,都找到相应物质的峰了,而且峰型挺不错的。请问大家在硝基呋喃的前处理过程中要注意什么问题。
[size=16px]源内裂解:[/size][font=Arial][size=16px][color=#4a90e2][/color][/size][/font][size=16px]当离子从高压电离源进入质量分析仪的真空区域时,可能发生离解或碎片化事件。某些药物代谢物的源内[back=#f7f8fa]([/back][font=Arial][back=#f7f8fa]CID[/back][/font][back=#f7f8fa])[/back]可能会产生与药物母体离子(目标分析物)相同的碎片离子。因此,将在用于定量药物的相同单反应监测(SRM)转换中检测到代谢物。在母体药物和代谢物之间缺乏足够的色谱分离度的情况下,可能会将代谢物来源中的CID产物离子误解为药物,从而使测定法没有选择性。[/size][size=16px]可以通过源内CID影响母体药物定量的最常见代谢物是:[/size][size=16px][color=#0080ff][b]酰基葡糖醛酸苷,O-和N-连接的葡糖醛酸苷,N-氧化物,硫酸盐结合物和内酯/羟基酸[/b][/color][/size][size=16px]如何控制或减少代谢产物的内源裂解:[/size][font=Arial][size=16px][back=#f7f8fa][/back][/size][/font][list][*][font=等线][size=16px]一般认为ESI应优先于大气压化学电离(APCI),以减少内源CID或化合物的热分解[/size][/font][*][font=等线][size=16px]ESI源的锥孔电压参数与源温度对源内裂解起主要作用[/size][/font][*][font=等线][size=16px]不同的加和离子,+NH4,以及负离子模式(-H),优于正离子模式(+H)[/size][/font][/list][font=等线][/font][size=18px][b][color=#ff0000]解决问题的终极方法,还是需要 裂解峰与待测物峰 色谱分离,其它的只能说是降低源内裂解发生的机率[/color][/b][/size][size=18px][b]文章来源,微信公众号“临床与分析哪些事”。[/b][/size][size=18px][b][color=#ff0000][/color][/b][/size]
近年来,随着质谱技术以及联用接口技术特别是包括电喷雾电离和大气压化学电离在内的大气压电离接口技术的突破,串联质谱及[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]技术在药物及其代谢物的定量和定性研究中发挥了极其重要的作用。对于药物的定量研究而言,常用的质谱为串联四极杆质谱,利用其多级离子选择的特殊性质,在多级离子选择监测扫描方式下,在保证质谱高灵敏度的同时,能极大的提高分析方法的特异性,减少或消除样品中无关物质的干扰,使得痕量分析和鉴定成为可能。并简化了生物样品的制备和分离过程,大大加速样品分析速度,特别适合对分析速度要求较高的药物筛选和临床试验生物样品的测定。在药物及其代谢物的定性研究中,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]系统中常用的质谱包括串联四极杆、离子阱、四极杆-飞行时间质谱等。这些质谱各有特点,结合同位素标记等技术,在药物代谢产物的分离鉴定、体内代谢途径确定等方面起到了别的分析技术无法替代的作用[1]。由于串联质谱及[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]技术的优越性,目前已广泛应用于药代动力学和药物代谢研究中。本文将重点介绍近年来色谱质谱联用技术在药代动力学和药物代谢研究中的应用。1 串联质谱技术在现代药代动力学研究中的应用1.1 在现代药代动力学研究中的作用 当今新药的研制和开发要求药代动力学研究向高通量和痕量检测的方向发展,串联质谱以其技术上的卓越性能在今天的药代动力学研究中发挥着重要的作用,推动了药代动力学研究的发展。1.1.1 极大提高了分析方法的灵敏度利用LC -MS 法成功建立了Beagle 犬血浆中人参皂苷20(R )-Rh 2的定量方法,并进行了药代动力学研究,结果测得20(R)-Rh 2在Beagle 犬血浆中最低定量限为015ng Πm L ,20(R )-Rh 2在015~200ng Πm L 浓度范围内线性关系良好(r 2=019998)。1.1.2 提高了分析方法的特异性和选择性 传统的分析方法缺点之一在于检测的特异性差,选择性差。通过串联质谱的分子离子和其特征碎片离子的质谱色谱图能分别进行定量,且定量的结果十分可靠。王洪允等以LC -MS ΠMS 同时测定了血浆中奥马曲拉及其中代谢物以及5中同位素内标,共10种化合物[3]1.1.3 实现药代动力学研究的高通量化,提高工作效率 在进行临床药代动力学研究时,经常需要在规定的时间内完成大批量生物样品的制备和分析。由于串联质谱固有的高灵敏度和特异性,能从复杂的生物基质中选择性地测定目标待测物,因此大大简化了样品制备和分离过程。另外,串联质谱对许多新技术表现出的兼容性,使得串联质谱在PK 研究的高通量化上表现出了极大的应用价值。这些技术包括在线固相萃取、柱切换技术和混合功能离子检测技术等。1.2 在药代动力学研究中的定量分析及应用1.2.1[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff] LC [/color][/url]-MS ΠMS 方法建立的基本流程 建立用于体内药物定量分析的LC -MS ΠMS 方法时,首先应该了解药物在生物基质中的特点。人体(或动物)在接受药物后,药物经吸收、分布、代谢(生物转化)、排泄等过程后,在体液中有浓度低,代谢复杂且代谢产物多等特点。因此,建立体内药物的定量方法时,其基本流程为()优化质谱参数建立合适的质谱条件以提高检测的灵敏度和特异性 (2)优化色谱系统建立最优化的色谱系统以实现更有效的分离 (3)优化样品制备方法以从复杂的生物基质中有效提纯药物。1.2.2 [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]ΠMS方法在药代动力学研究的应用 王宝莲[4]等采用高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联线性离子阱质谱同时检测五味子提取物中四种主要成分,采用电喷雾离子源,多反应监测模式进行检测。结果血浆样品经甲醇沉淀、高速离心后进行分析,各成分在0101~210μgΠm L的浓度范围内线性关系良好,最低定量限为0101~ 0102μgΠm L,建立了专属性强、快速、灵敏、可靠,可满足五味子提取物中四种主要成分药代动力学研究要求的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]ΠMS方法。赵建波等[5]利用高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱检测方法,研究秋水仙碱片的人体药动学。血浆样品011m L,经乙醚-二氯甲烷萃取,以ZOR BAX Ex2 tend-C18为色谱柱,流动相为甲醇-10mm olΠL乙酸铵,流速为111m LΠm in 采用质谱电喷雾离子化法,正离子多重反应检测(MRM)。结果秋水仙碱浓度在0105~10μgΠL范围内线性关系良好 平均回收率分别为(92147±1173)% 日内RS D≤2199%,日间RS D≤2122%,建立了适用于人血浆秋水仙碱浓度的测定及其药代动力学研究的简便、快速、准确可靠的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]方法。杨润涛等[6]建立同时测定大鼠血浆中白藜芦醇苷及其代谢产物白藜芦醇的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱方法。以Lichrospher C18色谱柱为分析柱,乙腈-水为流动相,采用电喷雾离子源(ESI),以多反应监测(MRM)模式检测,内标法定量,用于定量分析的离子反应分别为mΠz389Π227(白藜芦醇苷)和mΠz227Π143 (白藜芦醇)。在选定的样品预处理、色谱及质谱条件下,白藜芦醇苷、白藜芦醇及内标物能够达到基线分离而且离子化效果好。用LCΠMSΠMS法检测大鼠血浆中的白藜芦醇苷及其代谢产物白藜芦醇,线性范围014 ~200μgΠL。2 联质谱技术在药物代谢研究中的应用2.1 确证代谢物结构的基本步骤 药物在体内经过生物转化,形成的多数代谢产物保留了原药分子的基本骨架和部分亚结构,因此,代谢产物与原药可能具有相似的裂解规律,丢失一些相同的中性碎片或产生一些类似的特征离子。用串联质谱对原药和代谢物分别进行目离子扫描、子离子扫描和中性丢失扫描,即可迅速找到可能的代谢产物,并推导出大致结构。常雁等[7]总结了利用MSΠMS鉴定药物代谢物的方法,主要包括以下几个步骤:①测定原药的质谱 ②测定原药的子离子谱,选择质子化分子离子、加合离子和主要的碎片离子进行裂解 ③选择原药的主要中性丢失测定生物样品的中性丢失谱。图谱中的离子即为原药和可能的代谢物的分子离子 ④选择主要的子离子测定生物样品的母离子谱,所得母离子即为各个代谢物 ⑤测定生物样品中所有可能代谢物的子离子谱,解谱得到代谢物的结构 ⑥测定代谢物的子离子谱,选择任一新出现的中性丢失和子离子重复进行步骤3,4。2.2 在药物代谢产物结构确证中的应用 赵宇峰等[8]采用人肠内细菌和乌头碱体外温孵的方法,探讨乌头碱的代谢产物16-O-去甲基去氧乌头碱在人肠内的生物转化。利用离子阱电喷雾串联质谱(ESI MSΠMSn)方法直接分析16-O-去甲基去氧乌头碱的代谢产物。乌头类生物碱在ESI正离子模式条件下形成质子化分子[M+H]+。结果成功检测并鉴定了16-O-去甲基去氧乌头碱被人肠内细菌转化,通过脱乙酰基、脱苯甲酰基、脱甲基、脱羟基以及酯化反应产生新型的单酯型、双酯型和脂类生物碱等10余种代谢产物。马海英等[9]用电喷雾质谱(E SI-MS)法检测观察大鼠肠内菌离体对黄山药总皂苷(TS DP)的代谢及整体给予TS DP后吸收入血成分鉴定。结果显示TS DP容易被大鼠消化道菌群代谢,随着代谢时间的延长,出现了各种甾体皂苷的降解产物及终产物薯蓣皂苷元(D io)。赵宇峰等[10]利用离子阱和傅立叶变换离子回旋共振电喷雾串联质谱方法对牛蒡苷元和人肠内细菌真杆菌体外温孵样品进行测定,探讨牛蒡苷元的生物转化机理。木脂素类化合物在ESI负离子模式条件下形成准分子离子[M-H]-,在真杆菌的作用下,牛蒡苷元经过3次脱甲基反应最终生成4′,4″-二羟基肠内酯。3 展望串联质谱及[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]技术结合了色谱、质谱两者的优点,将色谱的高分离性能和质谱的高鉴别特点相结合,组成了较完美的现代分析技术,近年来在生物医学、药学、环境检测、毒物分析等领域发挥了巨大作用。且随着现代化高新技术的不断发展及[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱联用技术自身的优点,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱联用技术必将在未来几年不断发展且在药物分析中发挥越来越重要的作用。
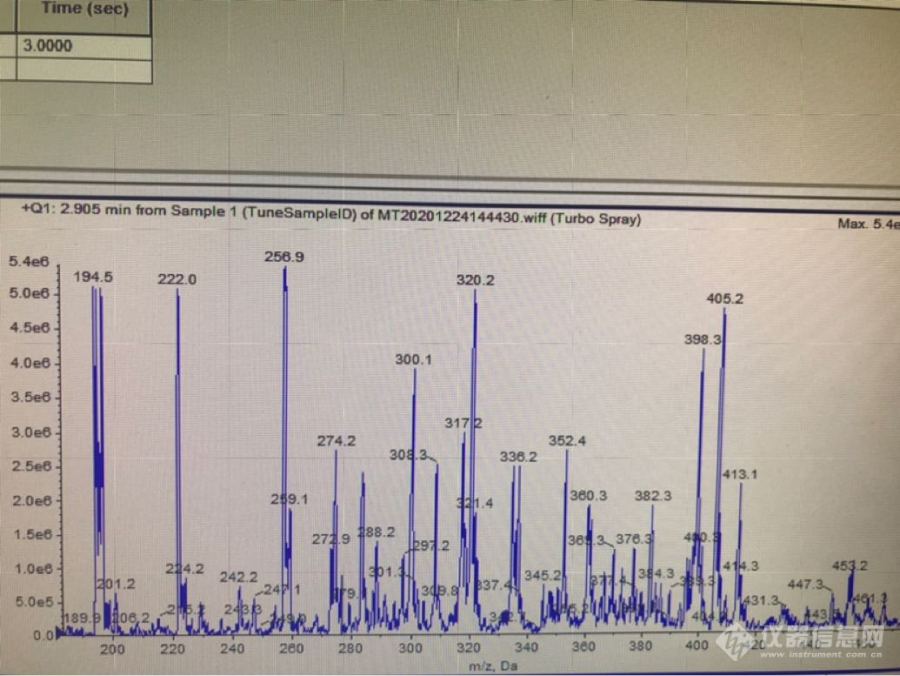
请教各位老师,小弟最近在参考国标用AB4000[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做硝基呋喃代谢物,按照国标的前处理方法,取了1ml 20ppm的标液,加了5ml盐酸和0.200ml 2硝基苯甲全,衍生过夜用甲醇稀释后优化质谱条件,找不到各个代谢物的衍生后母离子,又把衍生试剂加大了1ml,过夜,还是找不到,有做过的老师多多指点,谢谢!全扫描一级图如下[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/12/202012241454166709_6776_3557119_3.png[/img]
最近准备开验肉类中喹乙醇代谢物的检测项目,前后做了快两个月了,回收率始终不好,最好的也就50-60%方法试过:GB/T 20746-2006 牛、猪的肝脏和肌肉中卡巴氧和喹乙醇及代谢物残留量的测定 液相色谱-串联质谱法农业部1077号公告-5-2008 水产品中喹乙醇代谢物残留量的测定 高效液相色谱法以及一些文献方法。只有用乙腈提取的回收率有时可以,但是因为提取液含水浓缩过程很长,同时也影响最后的定量结果。现在头都大了,时间紧迫。各位大侠有没有靠谱的前处理方法啊,分享一下啊。先谢谢各位了!







 我要推广仪器
我要推广仪器
 下载APP
下载APP