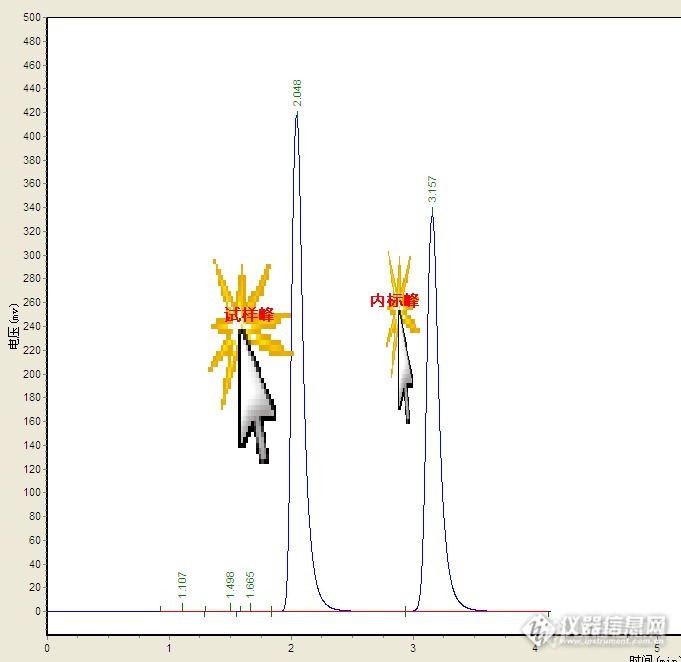
各位大佬,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]内标法,做内标标准曲线,是将等量的内标物加入到等体积不同浓度的标准物质中去,然后进色谱检验吗?
请问许多文献上提出核磁内标法(绝对测量法)定量分析时不需引进任何校正因子,不需制作标准曲线,但高效液相色谱为何需制作标准曲线?高效液相色谱和核磁内标法定量的原理差不多呀?[em09]
灭多威原药的分析方法一:灭多威的其他名称、结构式和基本物化参数如下:ISO通用名称:MethomylCIPAC数字代码;264化学名称:1-(甲硫基)亚乙基氨甲基氨基甲酸酯结构式 :CH3NH-CO-O-N=C-SCH3 CH3经验式:C5H10N2O2S相对分子质量:162.2(按1989年国际相对原子质量计)生物性质:内吸性杀虫剂熔点:78-79oC蒸汽压:(25oC):666.6X10-5Pa溶解度:(g /L25oC):水58 g /L,甲醇1000 g /L,丙酮730 g /L,乙醇420 g /L。稳定性:固体状态稳定,中性和微酸性介质中稳定,碱性溶液中缓慢分解,潮湿土镶中易分解。二:方法概要灭多威含量采用高效液相色谱法测定,UV检测器,254nm波长,使用乙酰苯胺内标物,内标面积法定量。三:鉴别试验:液相色谱法,在相同的色谱操作条件下,试样溶液主峰的保留时间与标样溶液色谱峰的保留时间,其相对偏差在1.5%以内,灭多威原药进行反相液相色谱测定.。四:仪器高效液相色谱仪:UV检测器,高压输液泵色谱柱:C18高效液相反相色谱柱过滤器:滤膜孔径:0.45nm进样针:50ul色谱工作站:高效液相色谱工作站五:试剂乙腈:色谱纯蒸馏水:双重蒸馏水,0.45um水膜过滤。灭多威标样:纯度:99.0%(m/m)内标溶液:精确称取5g乙酰苯胺内标物,放入干净的烧杯中,加入95g乙二醇,加热溶解,冷却备用。六:色谱操作条件流动相:乙腈/水=100/400,每500ml加入5滴磷酸调PH值,超声30min备用。稀释液:乙腈/水=100/400,超声30min备用。波长:254nm流速:1.00ml/min[size=11.0p
液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的外标法和内标定量有何区别?是如何处理的?
有内标物时高效液相色谱法中如何计算含量
【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,0.025 mol/L磷酸溶液-乙腈(87:13)用三乙胺调节pH值至3.0为流动相,检测波长为277nm。理论板数按环丙沙星峰计算应不低于2000,环丙沙星峰与内标物质峰的分离度应符合要求,拖尾因子应不大于2.5。内标溶液的制备 取恩诺沙星对照品25 mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,即得。测定法 取乳酸环丙沙星对照品约25 mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀;精密量取该溶液与内标溶液各5ml,置25ml量瓶中,用流动相稀释至刻度,摇匀,取20µ l注入液相色谱仪,记录色谱图;另取本品适量,同法测定。按内标法以峰面积计算,结果与0.7863相乘,即得。
请问一下,做高效液相色谱的内标法,做的是糖的色谱,测得果糖,山梨醇,葡萄糖,蔗糖这四个糖,不知道用哪一种内标物,还有就是使用了内标物之后怎么计算啊?
[b]高效液相色谱法的计算方法[/b]高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。 1.对仪器的一般要求 所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。 在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。 正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变, 以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。 2.系统适用性试验 按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子. (1) 色谱柱的理论板数(n) 在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。 (2) 分离度 定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。 (3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。 3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。 (2) 主成分自身对照法 当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。 (3) 内标法测定供试品中杂质的总量限度 采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。 如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。 (4) 内标法加校正因子测定供试品中某个杂质或主成分含量 按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。 再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。 当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。 (5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。[em61]
高效液相色谱法的计算方法高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。1.对仪器的一般要求所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。2.系统适用性试验按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子.(1) 色谱柱的理论板数(n)在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。(2) 分离度定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。(3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。(2) 主成分自身对照法当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。(3) 内标法测定供试品中杂质的总量限度采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。(4) 内标法加校正因子测定供试品中某个杂质或主成分含量按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。(5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。
液相色谱法中,内标法定量分析。有无要求内标物的峰面积和被测物的峰面积一样大小?有没有一定的范围要求?
我们用的是安捷伦1100的高效液相色谱仪,在做一个软膏剂的时候,标准规定的浓度进样10μl,峰形有前延峰,把浓度稀释一半,进样10μl,峰的对称性在1.0左右,如果再把浓度稀释一半,进样20μl的话,峰形又不对称了。大家分析一下是什么原因?液相色谱的进样量与峰形之间的关系怎样?(药品名字是“复方地塞米松乳膏”,内标法测定地塞米松含量,内标物是甲睾酮)
药典液相色谱方法的调整 • 根据药典附录“液相色谱法”规定,可调整适当参数 ---调整目的:满足系统适用性的要求 ---系统适用性的要求 ---HPLC方法调整的考虑因素 05版药典的系统适用性要求1、理论塔板数: ----反映整个色谱系统的状态 填料状态 管线连接 ----有不同的计算方法 主要是峰宽取值方法不同 不同计算方法计算结果有差异 ----影响因素: 被测组分的保留时间、进样量等 积分参数 系统死体积 测定色谱方法、样品与计算方法保持恒定,以便比较 05版药典的系统适用性要求 2、分离度: ---影响因素: * 影响柱效的因素 色谱柱尺寸 填料性能 进样量 * 影响分离选择性的因素 流动相组成 色谱柱品牌 柱温 * 柱外体积 ---有不同的计算方法,结果有差异 05版药典的系统适用性要求3、重复性(进样精密度): * 外标法:对照品溶液(n:5) 峰面积RSD:2.0% * 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标 溶液,分别至少进样2次,计算平均校正因子([font=Times New Roman
各位好,我刚学习液相,不是很明白在做液相时内标法和外标法,哪位能详细解释下啊。谢谢~~~
药典液相色谱方法的调整 • 根据药典附录“液相色谱法”规定,可调整适当参数 ---调整目的:满足系统适用性的要求 ---系统适用性的要求 ---HPLC方法调整的考虑因素 05版药典的系统适用性要求1、理论塔板数: ----反映整个色谱系统的状态 填料状态 管线连接 ----有不同的计算方法 主要是峰宽取值方法不同 不同计算方法计算结果有差异 ----影响因素: 被测组分的保留时间、进样量等 积分参数 系统死体积 测定色谱方法、样品与计算方法保持恒定,以便比较 05版药典的系统适用性要求 2、分离度: ---影响因素: * 影响柱效的因素 色谱柱尺寸 填料性能 进样量 * 影响分离选择性的因素 流动相组成 色谱柱品牌 柱温 * 柱外体积 ---有不同的计算方法,结果有差异 05版药典的系统适用性要求3、重复性(进样精密度): * 外标法:对照品溶液(n:5) 峰面积RSD:2.0% * 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标 溶液,分别至少进样2次,计算平均校正因子([font=Times New Roman
用内标法和外标法,液相色谱上机后的回收率是怎么计算的,我只知道定性就是看保留时间,可是要来定量我就不知道怎么算了。
高效液相色谱法与超高效液相色谱法的区别?
中药是一个典型的复杂未知样品,其化学物质基础的阐明是中药现代化的关键科学问题之一。液相色谱是中药分析中应用最普遍的技术,也是进一步与质谱、核磁共振等联用的基本分离手段。本论文在总结了中药复杂体系的特点和分析中需要解决的问题后,提出了中药分析的思路,开展了线性梯度下的液相色谱保留值规律、峰形规律的研究,发展了快速获取液相色谱保留参数、峰形参数的方法,从而将未知样品快速转化为色谱已知样品。并以实际样品藏茵陈和黄芪为例,应用发展的方法建立了相应的液相色谱参数数据库,提供中药活指纹图谱的信息基础。同时,面向实际应用,发展了中药组分的目标优化方法。建立了液-质联用表征黄芪皂甙的方法,建立了高效液相制备色谱快速制备黄芪组分的方法并初步发现了具有诱导分化白血病细胞的组分。发展了用五次线性梯度快速、准确地获取液相色谱保留值方程的方法。以线性梯度a、c 值预测任意梯度下的溶质的保留时间,并发展了基于内标峰的校正方法。应用于藏茵陈和黄芪样品的色谱保留参数获取,对c-a 关系进行了考察。基于EMG模型和自动曲线拟合技术,首次建立了以线性梯度快速获取液相色谱液相色谱峰形参数和峰形规律的方法。以线性梯度峰形规律预测了任意梯度条...[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31320]中药高效液相色谱分析方法发展的理论基础研究[/url]
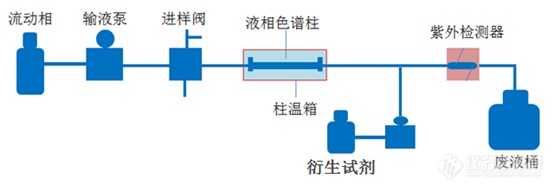
[align=center][size=21px][/size][/align][align=center][font=宋体]液相色谱衍生化法[/font][/align][font=宋体] 衍生化是一种利用化学变换把化合物类似化学结构或具有某种特性的物质的方法。通过衍生化法可以使样品中不易分离或分析的物质易于分离、分析。衍生化过程是使样品与衍生试剂在特定的设备内均匀混合,并在特定的条件下(如特定的温度、压力、光照等)充分反应。[/font][font=宋体] 在色谱分析中衍生化法一般有前处理衍生、柱前衍生、柱中衍生、柱后衍生等,液相色谱中柱后衍生应用较多,被称为柱后衍生液相色谱分析法。[/font][align=center][img=,555,191]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090917333555_9481_2369266_3.png!w555x191.jpg[/img][/align][img]file:///C:/Users/Administrator/AppData/Local/Temp/msohtmlclip1/01/clip_image002.jpg[/img][font=宋体] 衍生化常用的反应有酯化反应、酰化反应、硅烷化反应、环化反应、紫外衍生化反应、荧光衍生化反应等,通过给被测物加入衍生化试剂,再在加热等特定条件下使其发生衍生化反应。比如甲醛直接用液相色谱检测效果不好,而甲醛与[/font]2,4-[font=宋体]二硝基苯肼反应生成红色的[/font] 2,4-[font=宋体]二硝基苯腙,[/font]2,4-[font=宋体]二硝基苯腙用液相色谱检测效果就好的多。呋喃丹、甲萘威等农药成分用液相色谱检测效果不好,衍生化后用高效液相色谱荧光检测器检测效果很好。[/font][font=宋体] 像黄曲霉菌类,如黄曲霉毒素[/font]B1[font=宋体]在紫外光照射下可发出特定荧光,该类物质可以用特定的紫外光照射衍生化荧光检测器检测。[/font][font=宋体] 试剂衍生有的物质需要一种试剂一步衍生,有的物质需要多种试剂多布衍生,那衍生设备也就有单泵单衍生器和多泵多衍生器(通常是双泵双衍生器)不同配置,不管是单泵单衍生器还是多泵多衍生器现在大家基本都集成到一台设备里了。[/font][font=宋体] 美国[/font]Pickering[font=宋体]柱后衍生仪在国内占了很大一部分市场,国内柱后衍生仪主要厂家有天津奥特赛恩斯仪器有限公司、苏州华美辰、北京创新通恒等。[/font][align=center][img=,690,334]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090917557087_1544_2369266_3.png!w690x334.jpg[/img][img=,334,613]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090918005083_2140_2369266_3.png!w334x613.jpg[/img][/align] [font=宋体]随着发展,需要检测的物质会越来越多,液相色谱衍生化法应用的也会越来越多,衍生化法还会有较大发展空间。[/font][size=16px][/size]
乙腈:水=28:72时13-15min出峰的适合当液相色谱内标的化合物如题,在找一个化合物作为内标。液相色谱条件:菲罗门C18 250mm的柱子,C18预柱,30℃的柱温,流速1ml/min,流动相乙腈:水=28:72。看文献里貌似泼尼松龙比较合适,但是各种订货订不到,所以在寻找其他能做内标的化合物。
是指具有操作简便、分离速度快、分离效率高和检测灵敏度高等优良性能的液相色谱体系。液相色谱法早在1903年就由俄国植物学家Tswett发明,但早期的液相色谱法(古典液相色谱)柱效低、分离时间长,难以解决复杂样品的分离。到了20世纪60年代中后期,粒度小而均匀、传质速率快的色谱填料相继出现,使柱效显著提高,高压输液泵的使用解决了流动相流速慢的问题。从此液相色谱有了飞跃的发展,为区别于古典液相色谱法而称高效液相色谱法。HPLC几乎可以分离和分析任何物质,是最有效和应用最广泛的分离分析技术。
[color=#444444]最近想把液相色谱的外标法改为内标法,来克服进样量的要求,但液相内标物不是像[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的好选,求各位常用作反向液相内标物的物质都有哪些。是不是选择样品的同系物来做内标物[/color]
第一节 概 述 高效液相色谱法是继气相色谱之后,70年代初期发展起来的一种以液体做流动相的新色谱技术。 高效液相色谱是在气相色谱和经典色谱的基础上发展起来的。现代液相色谱和经典液相色谱没有本质的区别。不同点仅仅是现代液相色谱比经典液相色谱有较高的效率 和实现了自动化 操作。经典的液相色谱法,流动相在常压下输送,所用的固定相柱效低,分析周期长。而现代液相色谱法引用了气相色谱的理论,流动相改为高压输送(最高输送压力可达4.9107Pa);色谱柱是以特殊的方法用小粒径的填料填充而成,从而使柱效大大高于经典液相色谱(每米塔板数可达几万或几十万);同时柱后连有高灵敏度的检测器,可对流出物进行连续检测。因此,高效液相色谱具有分析速度快、分离效能高、自动化等特点。所以人们称它为高压、高速、高效或现代液相色谱法。 二、液相色谱分离原理及分类 和气相色谱一样,液相色谱分离系统也由两相——固定相和流动相组成。液相色谱的固定相可以是吸附剂、化学键合固定相(或在惰性载体表面涂上一层液膜)、离子交换树脂或多孔性凝胶;流动相是各种溶剂。被分离混合物由流动相液体推动进入色谱柱。根据各组分在固定相及流动相中的吸附能力、分配系数、离子交换作用或分子尺寸大小的差异进行分离。色谱分离的实质是样品分子(以下称溶质)与溶剂(即流动相或洗脱液)以及固定相分子间的作用,作用力的大小,决定色谱过程的保留行为。 根据分离机制不同,液相色谱可分为:液固吸附色谱、液液分配色谱、化合键合色谱、离子交换色谱以及分子排阻色谱等类型。三、液相色谱与气相色谱的比较 液相色谱所用基本概念:保留值、塔板数、塔板高度、分离度、选择性等与气相色谱一致。液相色谱所用基本理论:塔板理论与速率方程也与气相色谱基本一致。但由于在液相色谱中以液体代替气相色谱中的气体作为流动相,而液体和气体的性质不相同;此外,液相色谱所用的仪器设备和操作条件也与气相色谱不同,所以,液相色谱与气相色谱有一定差别,主要有以下几方面:(1)应用范围不同 气相色谱仅能分析在操作温度下能气化而不分解的物质。对高沸点化合物、非挥发性物质、热不稳定化合物、离子型化合物及高聚物的分离、分析较为困难。致使其应用受到一定程度的限制,据统计只有大约20%的有机物能用气相色谱分析;而液相色谱则不受样品挥发度和热稳定性的限制,它非常适合分子量较大、难气化、不易挥发或对热敏感的物质、离子型化合物及高聚物的分离分析,大约占有机物的70 ~ 80%。 (2)液相色谱能完成难度较高的分离工作 因为: ①气相色谱的流动相载气是色谱惰性的,不参与分配平衡过程,与样品分子无亲和作用,样品分子只与固定相相互作用。而在液相色谱中流动相液体也与固定相争夺样品分子,为提高选择性增加了一个因素。也可选用不同比例的两种或两种以上的液体作流动相,增大分离的选择性。 ②液相色谱固定相类型多,如离子交换色谱和排阻色谱等,作为分析时选择余地大;而气相色谱并不可能的。 ③ 液相色谱通常在室温下操作,较低的温度,一般有利于色谱分离条件的选择。(3)由于液体的扩散性比气体的小105倍,因此,溶质在液相中的传质速率慢,柱外效应就显得特别重要;而在气相色谱中,柱外区域扩张可以忽略不计。(4)液相色谱中制备样品简单,回收样品也比较容易,而且回收是定量的,适合于大量制备。但液相色谱尚缺乏通用的检测器,仪器比较复杂,价格昂贵。在实际应用中,这两种色谱技术是互相补充的。综上所述,高效液相色谱法具有高柱效、高选择性、分析速度快、灵敏度高、重复性好、应用范围广等优点。该法已成为现代分析技术的重要手段之一,目前在化学、化工、医药、生化、环保、农业等科学领域获得广泛的应用。
[color=#444444]目前正在做哌虫啶的体内代谢,现在遇到的问题是,如何选择一个合适的内标用作液相分析。它的结构式是 [/color][color=#444444]1-((6-氯吡啶-3-基)甲基)-5-丙氧基-7-甲基-8-硝基-1,2,3,5,6,7-六氢咪唑并吡啶 [/color][color=#444444]熔点: 130.2~131.9℃ [/color][color=#444444]蒸汽压(20℃) 200mPa [/color][color=#444444]溶解性 微溶于水,溶于二氯甲烷、氯仿、丙酮等大多数有机溶剂,[/color][color=#444444]色谱柱:Hypersil C18(4.6 mm × 250 mm,5 μm) [/color][color=#444444]流动相:乙腈-水(30:70),检测波长:354 nm,出峰时间为7.5min [/color][color=#444444]能否选择吡虫啉作为内标啊[/color]
液相色谱法 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]不能由色谱图直接给出未知物的定性结果,而必须由已知标准作对照定性。当无纯物质对照时,定性鉴定就很困难,这时需借助质谱、红外和化学法等配合。另外大多数金属盐类和热稳定性差的物质还不能分析。此缺点可高效液相色谱法来克服。液相色谱法就是用液体作为流动相的色谱法。1903 年俄国化学家M.C.茨维特首先将液相色谱法用于分离叶绿素。 原理和分类 液相色谱法的分离机理是基于混合物中各组分对两相亲和力的差别。根据固定相的不同,液相色谱分为液固色谱、液液色谱和键合相色谱。应用最广的是以硅胶为填料的液固色谱和以微硅胶为基质的键合相色谱。根据固定相的形式,液相色谱法可以分为柱色谱法、纸色谱法及薄层色谱法。按吸附力可分为吸附色谱、分配色谱、离子交换色谱和凝胶渗透色谱。近年来,在液相柱色谱系统中加上高压液流系统,使流动相在高压下快速流动,以提高分离效果,因此出现了高效(又称高压)液相色谱法。 品质软件试用下载:[URL=http://www.gztaiyou.com/jian/download.asp?instrument=315]http://www.gztaiyou.com/jian/download.asp?instrument=315[/URL] ①液固吸附色谱。高效液相色谱中的一种,是基于物质吸附作用的不同而实现分离。其固定相是一些具有吸附活性的物质如硅胶、氧化铝、分子筛、聚酰胺等。 ②液液分配色谱法。基于被测物质在固定相和流动相之间的相对溶解度的差异,通过溶质在两相之间进行分配以实现分离。根据固定相与流动相的极性不同,分为正相色谱和反相色谱。前者是用硅胶或极性键合相为固定相,非极性溶剂为流动相;后者是硅胶为基质的烷基键合相为固定相,极性溶剂为流动相,适用于非极性化合物的分离。 ③离子交换色谱法。基于离子交换树脂上可电离的离子与流动相中具有相同电荷的溶质离子进行可逆交换,依据这些离子对离子交换基具有不同的亲和力而实现分离。薄壳型离子交换树脂柱效高,主要用来分离简单的混合物;多孔性树脂进样容量大,主要用来分离复杂混合物。 ④凝胶渗透色谱法[1] 。又称为尺寸排阻色谱法 。1959年首先用于生物化学领域。以溶剂为流动相,多孔填料(如多孔硅胶、多孔玻璃)或多孔交联高分子凝胶为分离介质的液相色谱法。当混合物溶液入凝胶色谱柱后,流经多孔凝胶时,体积比多孔凝胶孔隙大的分子不能渗透到凝胶孔隙里去而从凝胶颗粒间隙中流过,较早地被冲洗出柱外,而小分子可渗透到凝胶孔隙里面去,较晚地被冲洗出来,混合物经过凝胶色谱柱后就按其分子大小顺序先后由柱中流出达到分离的目的。用凝胶渗透色谱的优点是:分离不需要梯度冲洗装置 ;同样大小的柱能接受比通常液相色谱大得多的试样量;试样在柱中稀释少,因而容易检测;组分的保留时间可提供分子尺寸信息;色谱柱寿命长。它的缺点是:不能分离分子尺寸相同的混合物,色谱柱的分离度低;峰容量小;可能有其他保留机理起作用时引起干扰。凝胶渗透色谱法为测定高聚物分子量和分子量分布提供了一个有效的方法,此外还可用来分离齐聚物、单体和聚合物添加剂等。 ⑤[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法。采用柱色谱技术的一种高效液相色谱法,样品展开方式采用洗脱法。根据不同的分离方式,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]可以分为高效[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url] 、离子排斥色谱和流动相[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]3类。高效[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法使用低容量的离子交换树脂,分离机理主要是离子交换。离子排斥色谱法用高容量的树脂,分离机理主要是利用离子排斥原理。流动相[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]用不含离子交换基团的多孔树脂,分离机理主要是基于吸附和离子对的形成。 [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]由淋洗液贮存器 、泵 、进样阀 、分离柱 、抑制柱、电导检导器和数据处理单元等组成。[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]最重要的部件是分离柱,装有离子交换树脂。抑制柱是抑制型[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]的关键部件,其作用是将淋洗液转变成低电导部分,以降低来自淋洗液的背景电导,同时将样品离子转变成其相应的酸或碱,以增加其电导。分离阴离子,抑制柱填充强酸性阳离子交换树脂;分离阳离子,抑制柱填充强碱性阴离子交换树脂。检测器分通用型检测器与专用型检测器。前者如电导检测器,对检测池中所有离子都有响应;后者如紫外-可见分光光度计,对离子具有选择性响应。 [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法具有快速、灵敏、选择性好和同时测定多组分的优点。尤其对于阴离子的测定,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]的出现是分析化学中的一项突破性的新进展。[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法主要用于测定各种离子含量,广泛应用于水、纸浆和漂白液、食品分析、生物体液、钢铁和环境分析等各个领域。 设备 高效液相色谱仪由输出泵、进样装置、色谱柱 、梯度冲洗装置、检测器及数据处理和微机控制单元组成。输出泵的功能是将冲洗剂在高压下连续不断地送入柱系统,使混合物试样在色谱中完成分离过程 。常用的进样方式有3种:注射器隔膜进样、阀进样和自动进样器进样。色谱柱的功能是将混合物中各组分分离。梯度冲洗又称溶剂程序,通过连续改变冲洗剂的组成,改善复杂样品的分离度,缩短分析周期和改善峰形,其功能类似于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中的程序升温。检测器的功能是将从色谱柱中流出的已经分离的组分显示出来或转换为相应的电信号,主要有紫外吸收检测器、荧光检测器、电化学检测器和折光示差检测器,其中以紫外吸收检测器使用最广。现代化的仪器都配有计算机,以实现自动处理数据、绘图和打印分析报告。
[align=center][b]液相色谱柱的发展历程[/b][/align]每次我们看到色谱柱的时候,都会想到明亮的不锈钢柱管,但是色谱柱柱管发展到今天也是历经坎坷的,科学技术的发展进步从来都是曲折的。1、玻璃柱管时代最早的色谱柱填料是填充在玻璃管里面的,玻璃管的机械强度,所以那个时代的色谱柱柱压不能太高,属于低压液相色谱的年代。这个是现代液相色谱分析的雏形,曾经的屠呦呦老先生靠最原始的柱层析提取出了青蒿素,这个是时代的进步!2、塑料柱管时代塑料取代玻璃是色谱技术的一大进步,塑料的工程强度要大于玻璃。所以用塑料装填色谱填料也流行一时。今天,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]的色谱柱柱管依然使用的是塑料。但是液相色谱柱却摒弃了塑料。液相色谱的流动相多为甲醇乙腈等有机溶剂,塑料可能与有机溶剂相互作用,甚至发生溶解。3、不锈钢柱管时代不锈钢色谱柱柱管的产生,意味着液相色谱的发展进入了一个崭新的时代。色谱柱可以耐更高的压力,所以通常我们说液相色谱是高效液相色谱或者高压液相色谱,其原因应该来源于此。我在网上看到,有的人说是当初液相色谱进入中国的时候,翻译错误造成的。但是本人认为从事物发展的脉络来看,叫高压液相似乎更加合理。随着填料技术的不断发展,液相色谱又被推陈出新到一个新的高度,超高压液相色谱。色谱柱的发展历程是色谱柱柱管的发展历程,也是色谱填料技术发展的历程,这两个因素相互作用,相互促进,才有了今天色谱柱技术的飞跃发展!我们在使用不锈钢色谱柱的时候,不应该忘记液相色谱柱的前世今生!尽管今天我们不会叫液相色谱仪为“高效液相色谱仪”!所以,我们说色谱柱是液相色谱的核心!
第一节 概 述 高效液相色谱法是继气相色谱之后,70年代初期发展起来的一种以液体做流动相的新色谱技术。 高效液相色谱是在气相色谱和经典色谱的基础上发展起来的。现代液相色谱和经典液相色谱没有本质的区别。不同点仅仅是现代液相色谱比经典液相色谱有较高的效率 和实现了自动化 操作。经典的液相色谱法,流动相在常压下输送,所用的固定相柱效低,分析周期长。而现代液相色谱法引用了气相色谱的理论,流动相改为高压输送(最高输送压力可达4.9107Pa);色谱柱是以特殊的方法用小粒径的填料填充而成,从而使柱效大大高于经典液相色谱(每米塔板数可达几万或几十万);同时柱后连有高灵敏度的检测器,可对流出物进行连续检测。因此,高效液相色谱具有分析速度快、分离效能高、自动化等特点。所以人们称它为高压、高速、高效或现代液相色谱法。 二、液相色谱分离原理及分类 和气相色谱一样,液相色谱分离系统也由两相——固定相和流动相组成。液相色谱的固定相可以是吸附剂、化学键合固定相(或在惰性载体表面涂上一层液膜)、离子交换树脂或多孔性凝胶;流动相是各种溶剂。被分离混合物由流动相液体推动进入色谱柱。根据各组分在固定相及流动相中的吸附能力、分配系数、离子交换作用或分子尺寸大小的差异进行分离。色谱分离的实质是样品分子(以下称溶质)与溶剂(即流动相或洗脱液)以及固定相分子间的作用,作用力的大小,决定色谱过程的保留行为。 根据分离机制不同,液相色谱可分为:液固吸附色谱、液液分配色谱、化合键合色谱、离子交换色谱以及分子排阻色谱等类型。三、液相色谱与气相色谱的比较 液相色谱所用基本概念:保留值、塔板数、塔板高度、分离度、选择性等与气相色谱一致。液相色谱所用基本理论:塔板理论与速率方程也与气相色谱基本一致。但由于在液相色谱中以液体代替气相色谱中的气体作为流动相,而液体和气体的性质不相同;此外,液相色谱所用的仪器设备和操作条件也与气相色谱不同,所以,液相色谱与气相色谱有一定差别,主要有以下几方面:(1)应用范围不同 气相色谱仅能分析在操作温度下能气化而不分解的物质。对高沸点化合物、非挥发性物质、热不稳定化合物、离子型化合物及高聚物的分离、分析较为困难。致使其应用受到一定程度的限制,据统计只有大约20%的有机物能用气相色谱分析;而液相色谱则不受样品挥发度和热稳定性的限制,它非常适合分子量较大、难气化、不易挥发或对热敏感的物质、离子型化合物及高聚物的分离分析,大约占有机物的70 ~ 80%。 (2)液相色谱能完成难度较高的分离工作 因为: ①气相色谱的流动相载气是色谱惰性的,不参与分配平衡过程,与样品分子无亲和作用,样品分子只与固定相相互作用。而在液相色谱中流动相液体也与固定相争夺样品分子,为提高选择性增加了一个因素。也可选用不同比例的两种或两种以上的液体作流动相,增大分离的选择性。 ②液相色谱固定相类型多,如离子交换色谱和排阻色谱等,作为分析时选择余地大;而气相色谱并不可能的。 ③ 液相色谱通常在室温下操作,较低的温度,一般有利于色谱分离条件的选择。(3)由于液体的扩散性比气体的小105倍,因此,溶质在液相中的传质速率慢,柱外效应就显得特别重要;而在气相色谱中,柱外区域扩张可以忽略不计。(4)液相色谱中制备样品简单,回收样品也比较容易,而且回收是定量的,适合于大量制备。但液相色谱尚缺乏通用的检测器,仪器比较复杂,价格昂贵。在实际应用中,这两种色谱技术是互相补充的。综上所述,高效液相色谱法具有高柱效、高选择性、分析速度快、灵敏度高、重复性好、应用范围广等优点。该法已成为现代分析技术的重要手段之一,目前在化学、化工、医药、生化、环保、农业等科学领域获得广泛的应用。第二节 高效液相色谱仪 高效液相色谱仪由 高压输液系统、进样系统、分离系统、检测系统、记录系统 等五大部分组成(图14-2)。 分析前,选择适当的色谱柱和流动相,开泵,冲洗柱子,待柱子达到平衡而且基线平直后,用微量注射器把样品注入进样口,流动相把试样带入色谱柱进行分离,分离后的组分依次流入检测器的流通池,最后和洗脱液一起排入流出物收集器。当有样品组分流过流通池时,检测器把组分浓度转变成电信号,经过放大,用记录器记录下来就得到色谱图。色谱图是定性、定量和评价柱效高低的依据。 一、高压输液系统高压输液系统由 溶剂贮存器、高压泵、梯度洗脱装置和压力表 等组成。 1.溶剂贮存器 溶剂贮存器一般由玻璃、不锈钢或氟塑料制成,容量为1到2 L,用来贮存足够数量、符合要求的流动相。 2.高压输液泵 高压输液泵是高效液相色谱仪中关键部件之一,其功能是 将溶剂贮存器中的流动相以高压形式连续不断地送入液路系统,使样品在色谱柱中完成分离过程。由于液相色谱仪所用色谱柱径较细,所填固定相粒度很小,因此,对流动相的阻力较大,为了使流动相能较快地流过色谱柱,就需要高压泵注入流动相。 对泵的要求:输出压力高、流量范围大、流量恒定、无脉动,流量精度和重复性为0.5%左右。此外,还应耐腐蚀,密封性好。 高压输液泵,按其性质可分为恒压泵和恒流泵两大类。 恒流泵是能给出恒定流量的泵,其流量与流动相粘度和柱渗透无关。 恒压泵是保持输出压力恒定,而流量随外界阻力变化而变化,如果系统阻力不发生变化,恒压泵就能提供恒定的流量。3. 梯度洗脱装置 梯度洗脱就是在分离过程中使两种或两种以上不同极性的溶剂按一定程序连续改变它们之间的比例,从而使流动相的强度、极性、pH值或离子强度相应地变化,达到提高分离效果,缩短分析时间的目的。 梯度洗脱装置分为两类: 一类是外梯度装置(又称低压梯度),流动相在常温常压下混合,用高压泵压至柱系统,仅需一台泵即可。 另一类是内梯度装置(又称高压梯度),将两种溶剂分别用泵增压后,按电器部件设置的程序,注入梯度混合室混合,再输至柱系统。 梯度洗脱的实质是通过不断地变化流动相的强度,来调整混合样品中各组分的k值,使所有谱带都以最佳平均k值通过色谱柱。它在液相色谱中所起的作用相当于气相色谱中的程序升温,所不同的是,在梯度洗脱中溶质k值的变化是通过溶质的极性、pH值和离子强度来实现的,而不是借改变温度(温度程序)来达到。二、进样系统 进样系统包括进样口、注射器和进样阀等,它的作用是把分析试样有效地送入色谱柱上进行分离。三、分离系统 分离系统包括色谱柱、恒温器和连接管等部件。色谱柱一般用内部抛光的不锈钢制成。其内径为2 ~ 6mm,柱长为10 ~50cm,柱形多为直形,内部充满微粒固定相。柱温一般为室温或接近室温。四、检测器 检测器是液相色谱仪的关键部件之一。对检测器的要求是:灵敏度高,重复性好、线性范围宽、死体积小以及对温度和流量的变化不敏感等。 在液相色谱中,有两种类型的检测器,一类是溶质性检测器,它仅对被分离组分的物理或物理化学特性有响应。属于此类检测器的有紫外、荧光、电化学检测器等;另一类是总体检测器,它对试样和洗脱液总的物理和化学性质响应。属于此类检测器有示差折光检测器等。第三节 高效液相色谱的固定相 和流动相一、固定相 高效液相色谱固定相以承受高压能力来分类,可分为刚性固体和硬胶两大类。 刚性固体以二氧化硅为基质,可承受7.0108~1.0109Pa的高压,可制成直径、形状、孔隙度不同的颗粒。如果在二氧化硅表面键合各种官能团,可扩大应用范围,它是目前最广泛使用的一种固定相。 硬胶主要用于离子交换和尺寸排阻色谱中,它由聚苯乙烯与二乙烯苯基交联而成。可承受压力上限为3.5108Pa。固定相按孔隙深度分类,可分为表面多孔型和全多孔型固定相两类。1. 表面多孔型固定相 它的基体是实心玻璃球,在玻璃球外面覆盖一层多孔活性材料,如硅胶、氧化硅、离子交换剂、分子筛、聚酰胺等。这类固定相的多孔层厚度小、孔浅,相对死体积小,出峰迅速、柱效亦高;颗粒较大,渗透性好,装柱容易,梯度淋洗时能迅速达到平衡,较适合做常规分析。由于多孔层厚度薄,最大允许量受到限制。2. 全多孔型固定相 由直径为10nm的硅胶微粒凝聚而成。这类固定相由于颗 粒很细(5~10m),孔仍然较浅,传质速率快,易实现高效、高速。特别适合复杂混合物分离及痕量分析。二、流动相 由于高效液相色谱中流动相是液体,它对组分有亲合力,并参与固定相对组分的竞争,因此,正确选择流动相直接影响组分的分离度。对流动相溶剂的要求是:(1)溶剂对于待测样品,必须具有合适的极性和良好的选 择性。(2)溶剂与检测器匹配。对于紫外吸收检测器,应注意选 用检测器波长比溶剂的紫外截止波长 要长。所谓溶剂的紫外截止波长指当小于截止波长的辐射通过溶剂时, 溶剂对此辐射产生强烈吸收,此时溶剂被看作是光学不 透明的,它严重干扰组分的吸收测量。 对于折光率检测器,要求选择与组分折光率有较
[color=#444444]用高效液相色谱检测血液样品中的一些物质,看到文献中有很多前处理的方法,请问血液的前处理有什么依据和需要注意的方面?还有,看到文献上都有加入一些内标物,请问内标物是否一定要加?谢谢[/color]

【作者】 方奕珊; 李来生; 陈红; 张杨;【Author】 FANG Yi-shan,LI Lai-sheng,CHEN Hong,ZHANG Yang(The Centre of Analysis and Testing,Nanchang University,Nanchang 330047,China)【机构】 南昌大学分析测试中心;【摘要】 以4-二甲氨吡啶为内标,发展了一种高效液相色谱法,用于快速测定鸡蛋中氯羟吡啶的残留量。鸡蛋样品中的氯羟吡啶经乙腈匀浆提取,然后用碱性氧化铝柱富集,甲醇洗脱液浓缩后用流动相溶解残渣,0.45μm滤膜过滤后进样分析。色谱柱采用Platisil-C18柱(4.6mm×250mm,5μm),流动相为0.05mol.L-1甲酸铵-乙腈(85:15,v/v,pH 6.4),流速为0.8mL.min-1,二极管阵列定量检测波长为267nm,柱温25℃,进样量20μL。氯羟吡啶在0.5~80μg.mL-1范围内呈现良好的线性关系,线性方程为Y=0.254 4 X+0.703 9,相关系数为0.999 6。3个不同水平的标准添加平均回收率分别为85.3%,88.2%和83.7%,相对标准偏差≤2.25%(n=4)。最低检出限为1.8mg.kg-1。该方法快速、简便、准确、重现性好,适用于鸡蛋中兽药氯羟吡啶残留量的快速检测。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301524_380581_2379123_3.jpg
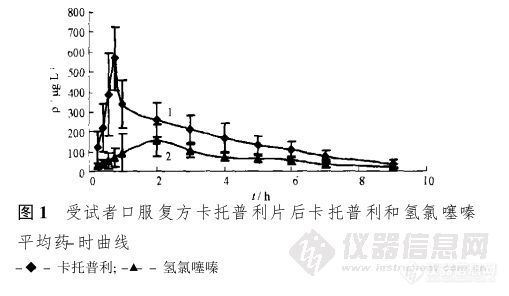
【作者】 黄滔敏; 何忠; 郑晓伟; 绍路平; 段更利; 【Author】 HUANG Tao-min~(1,2),HE Zhong~1,ZHENG Xiao-wei~1,SHAO Lu-ping~1,DUAN Geng-li~(1)(1.Department of Pharm-analysis,College of Pharmacy,Fudan University,Shanghai 200032,China;2.Department of Eye & ENT Hospital,Fudan University,Shanghai 200031,China) 【机构】 复旦大学药学院药物分析教研室; 复旦大学药学院药物分析教研室 上海200032; 复旦大学附属眼耳鼻喉科医院; 上海200031; 上海200032; 【摘要】 目的建立同时测定血浆中卡托普利和氢氯噻嗪浓度的高效液相色谱法,并将该方法应用到健康受试者口服复方卡托普利片后卡托普利和氢氯噻嗪血药浓度测定。方法采用Diamonsil C18(4.6 mm×150 mm,5μm)色谱柱,流动相:A:pH 1.8醋酸水溶液,B:乙腈,0~5 min,A-B=90∶10,8 min后,A-B=60∶40;柱温30℃,流速1.2 mL.min-1,检测波长263 nm,磺胺嘧啶为内标。卡托普利经对溴苯乙酰基溴(p-BPB)衍生化后同时测定卡托普利和氢氯噻嗪血药浓度。结果在一定浓度范围内,被测药物(卡托普利和氢氯噻嗪)分别和内标的峰面积之比与浓度成良好的线性关系,提取回收率均高于70%,日内和日间精密度均在合格范围内。结论本试验建立的方法简便、快速、准确,适用于复方卡托普利片剂中卡托普利和氢氯噻嗪体内血药浓度测定。 更多还原 【关键词】 高效液相色谱法; 血药浓度; 卡托普利; 氢氯噻嗪;http://ng1.17img.cn/bbsfiles/images/2012/09/201209022116_388007_1838299_3.jpg
wufeng的液相色谱仪,现在做内标样的峰高和峰面积是原来的2倍,冲过柱子了,有哪位知道是怎么回事吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP