推荐厂家
暂无
暂无
 白金2年
白金2年
 400-803-5889
400-803-5889
 留言咨询
留言咨询
留言咨询
留言咨询
 金牌12年
400-860-5168转2577
留言咨询
金牌12年
400-860-5168转2577
留言咨询
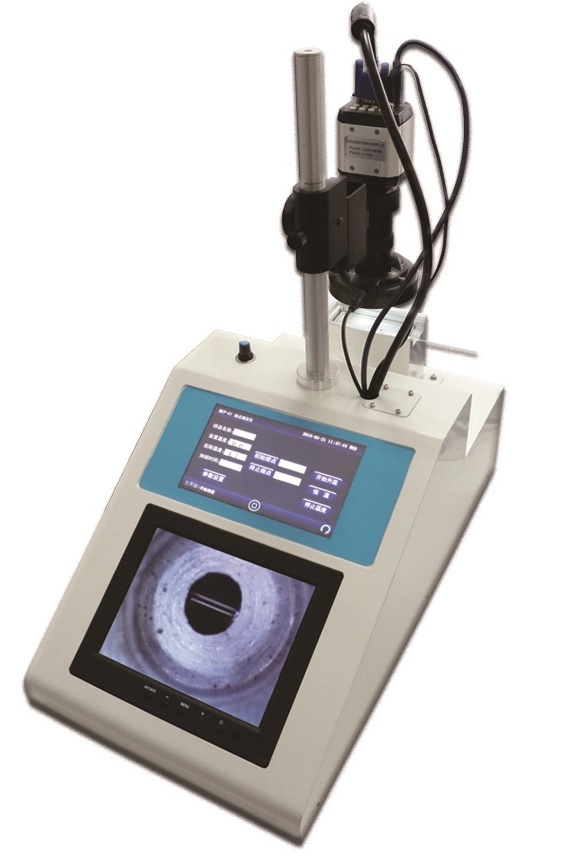 400-860-5168转3723
留言咨询
400-860-5168转3723
留言咨询
 400-825-5787
留言咨询
400-825-5787
留言咨询
 400-860-5168转3723
留言咨询
400-860-5168转3723
留言咨询






实验室有一台数显熔点仪,但有时样品测定的结果有偏差,特别是和供应商提供的检测报告误差有2度,有时在质量指标的边缘,很难判断。所以想购买一个老式的油浴装置,温度计已经买了,但市场上很难看到烧杯配加热管的装置,各位谁有看到,推荐一下,最好在南京市内。
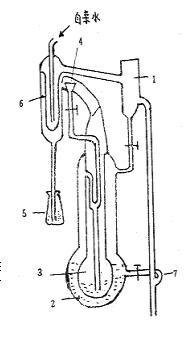
要做实验测定!1、熔点的测定化合物的熔点是指在常压下该物质的固—液两相达到平衡时的温度。但通常把晶体物质受热后由固态转化为液态时的温度作为该化合物的熔点。纯净的固体有机化合物一般都有固定的熔点。在一定的外压下,固液两态之间的变化是非常敏锐的,自初熔至全熔(称为熔程)温度不超过0.5-1℃。若混有杂质则熔点有明确变化,不但熔点距扩大,而且熔点也往往下降。因此,熔点是晶体化合物纯度的重要指标。有机化合物熔点一般不超过350℃,较易测定,故可借测定熔点来鉴别未知有机物和判断有机物的纯度。在鉴定某未知物时,如测得其熔点和某已知物的熔点相同或相近时,不能认为它们为同一物质。还需把它们混合,测该混合物的熔点,若熔点仍不变,才能认为它们为同一物质。若混合物熔点降低,熔程增大,则说明它们属于不同的物质。故此种混合熔点试验,是检验两种熔点相同或相近的有机物是否为同一物质的最简便方法。熔点装置图:2、沸点的测定液体的分子由于分子运动有从表面逸出的倾向,这种倾向随着温度的升高而增大,进而在液面上部形成蒸气。当分子由液体逸出的速度与分子由蒸气中回到液体中的速度相等,液面上的蒸气达到饱和,称为饱和蒸气。它对液面所施加的压力称为饱和蒸气压。实验证明,液体的蒸气压只与温度有关。即液体在一定温度下具有一定的蒸气压。当液体的蒸气压增大到与外界施于液面的总压力(通常是大气压力)相等时,就有大量气泡从液体内部逸出,即液体沸腾。这时的温度称为液体的沸点。通常所说的沸点是指在101.3kPa下液体沸腾时的温度。在一定外压下,纯液体有机化合物都有一定的沸点,而且沸点距也很小(0.5-1℃)。所以测定沸点是鉴定有机化合物和判断物质纯度的依据之一。测定沸点常用的方法有常量法(蒸馏法)和微量法(沸点管法)两种。 实验步骤1、熔点的测定毛细管法:①准备熔点管:将毛细管截成6~8cm长,将一端用酒精灯外焰封口(与外焰成40o角转动加热)。防止将毛细管烧弯、封出疙瘩。②装填样品:取0.1~0.2g预先研细并烘干的样品,堆积于干净的表面皿上,将熔点管开口一端插入样品堆中,反复数次,就有少量样品进入熔点管中。然后将熔点管在垂直的约40cm的玻璃管中自有下落,使样品紧密堆积在熔点管的下端,反复多次,直到样品高约2~3cm为止,每种样品装2~3根。③仪器装置:将b形管固定于铁架台上,倒入液体石蜡做为浴液,其用量以略高于b形管的上侧管为宜。将装有样品的熔点管用橡皮圈固定于温度计的下端,使熔点管装样品的部分位于水银球的中部。然后将此带有熔点管的温度计,通过有缺口的软木塞小心插入b形管中,使之与管同轴,并使温度计的水银球位于b形管两支管的中间。④熔点测定:粗测:慢慢加热b形管的支管连接处,使温度每分钟上升约5℃。观察并记录样品开始熔化时的温度,此为样品的粗测熔点,作为精测的参考。精测:待浴液温度下降到30℃左右时,将温度计取出,换另一根熔点管,进行精测。开始升温可稍快,当温度升至离粗测熔点约10℃时,控制火焰使每分钟升温不超过1℃。当熔点管中的样品开始塌落,湿润,出现小液滴时,表明样品开始溶化,记录此时温度即样品的始熔温度。继续加热,至固体全部消失变为透明液体时再记录温度,此即样品的全熔温度。样品的熔点表示为:t始熔~t全熔。实测:尿素(已知物,133~135℃)、桂皮酸(未知物,132~133℃),混合物(尿素-桂皮酸=1:1,100℃左右)。实验过程中,粗测一次,精测两次。2、沸点的测定微量法测定沸点:①沸点管的制备:沸点管由外管和内管组成,外管用长7~8厘米、内径0.2~0.3cm的玻璃管将一端烧熔封口制得,内管用市购的毛细管截取3~4cm封其一端而成。测量时将内管开口向下插入外管中。②沸点的测定:取1~2滴待测样品滴入沸点管的外管中(思考题9),将内管插入外管中,然后用小橡皮圈把沸点附于温度计旁,再把该温度计的水银球位于b形管两支管中间,然后加热。加热时由于气体膨胀,内管中会有小气泡缓缓逸出,当温度升到比沸点稍高时,管内会有一连串的小气泡快速逸出。这时停止加热,使溶液自行冷却,气泡逸出的速度即渐渐减慢。在最后一气泡不再冒出并要缩回内管的瞬间记录温度,此时的温度即为该液体的沸点,待温度下降15~20℃后,可重新加热再测一次(2次所得温度数值不得相差1℃)。按上述方法进行如下测定:CCl4沸点(76℃)。 注意事项1.熔点管必须洁净。如含有灰尘等,能产生4—10OC的误差。2.熔点管底未封好会产生漏管。3.样品粉碎要细,填装要实,否则产生空隙,不易传热,造成熔程变大。4.样品不干燥或含有杂质,会使熔点偏低,熔程变大。5.样品量太少不便观察,而且熔点偏低;太多会造成熔程变大,熔点偏高。6.升温速度应慢,让热传导有充分的时间。升温速度过快,熔点偏高。7.熔点管壁太厚,热传导时间长,会产生熔点偏高。
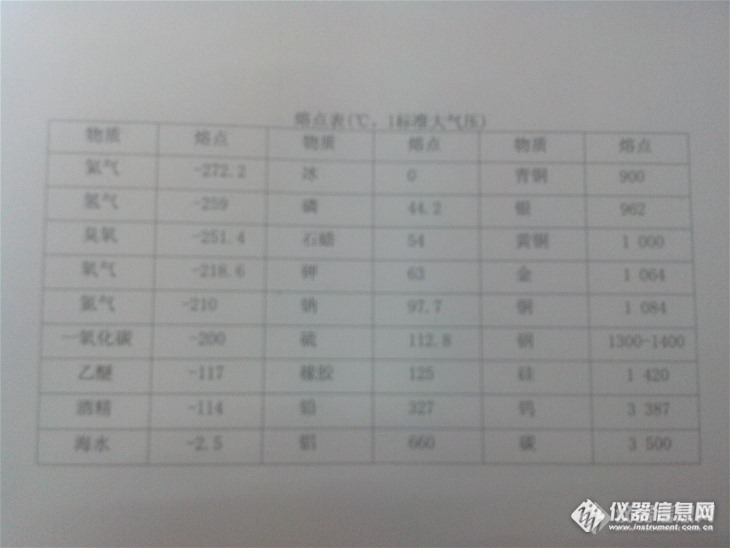
物质熔点测定方法物质的熔点,一般情况下就是指该物质固、液两相可以共存并处于平衡的温度,一说到这个,我们就会想到冰和水,以冰熔化成水为例,在一个大气压下冰的熔点是0℃,而温度为0℃时,冰和水可以共存在通常的情况下,物体在不同的温度下,其状态也是不同的,且即时在同一温度,其状态也是不确定的,如晶体处于熔点温度时,可能有三种状态,即可能固态、可能液态、也可能固液共存,常用物质溶解表如下:熔点表http://ng1.17img.cn/bbsfiles/images/2013/11/201311131143_476934_2154459_3.jpg那么上表这些物质的熔点怎么得来的的呢?有没有什么方法可以测试物质的熔点呢,那就看看吧!1.原理以加热的方式使毛细管中的试样从低于其初熔时的温度逐渐升至高于其终熔时的温度,通过目测分别观察其初熔和终熔温度,即为试样的初熔点和终熔点。2.仪器(1)毛细管:硬质玻璃毛细管,内径0.9~1.1mm,壁厚0.15~0.2mm,长110~120mm,一端封口。(2)测量温度计:分度值为0.1℃,长约400mm,全浸或局浸式并经校正的温度计,根据熔点范围选择适当量程。(3)辅助温度计:分度值为1℃的普通温度计,量程与测量温度计一致。(4)加热器:加热均匀、安全、易控制温度的加热装置。(5)熔点测定装置:容积250ml的内热式烧瓶,直径80mm,瓶长130~140mm,瓶口处有一温度计插口直径约10mm的圆孔,在瓶口上方有两个相对称的毛细管插口直径约2mm的圆孔。3.传热液体201型甲基硅油(粘度在500号以上),或硫酸与硫酸钾混合液(220ml浓硫酸中加入70~80g无水硫酸钾,再加入0.1~0.2g硝酸钾)。4.样品的干燥取少量混匀、研细的试样放于干燥、清洁的表面皿中,于105℃干燥箱中干燥30min后取出,放在干燥器中冷却备用。5.测定将少量干燥、研细的试样装入清洁、干燥、一端封口的毛细管中,取一高约800mm、直径10mm的干燥玻璃管直立于玻璃板上,将装有试样的毛细管经玻璃管投掷8~10次,直至毛细管内试样紧缩至3-4mm高,再将开口一端封熔,圆底烧瓶中注入其体积四分之三的传热液体,测量温度计插入试管内的传热液体中,并使温度计的中间泡也浸没于传热液体中,温度计不得碰及管底。加热使传热液体温度缓缓上升至熔点前10℃时,将装有试样的毛细管通过锥形导管插入试管内并附着于温度计水银球的中部,继续加热,调节加热器使温度上升速度为0.5~1.0℃/min。观察毛细管中试样熔化情况,当试样开始出现明显的局部液化现象时的温度为初熔点。6.结果的表示与计算若测定中使用的是局浸式温度计,初熔点t=t+△t;若测定中使用的是全浸式温度计,初熔点t按下式计算: t=t1+△t1+△t2+△t3△t1=0.00016h1(t1-t2)△t2=0.00016h2(t1-t3)式中 t——初熔点,℃△t1——温度计露出液面至塞口处的水银柱校正值,℃△t2——温度计露出塞外的水银柱校正值,℃△t3——温度计的示值校正值,℃t1——观察温度,t2——试管内液面至塞口的中间处的温度,t3[









 我要推广仪器
我要推广仪器
 下载APP
下载APP



