如题,有人支持持续更新[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=52001]熔点测定[/url]《741》熔距或熔点为了药典应用起见,除下述方法Ⅱ和方法Ⅲ另有规定外,固体的熔点或熔距一般指固体融合并完全融化的温度范围或温度。可以采用具有同样精度的任何装置或方法进行测定。其准确度应定期用六种美国药典的熔点参比标准品中的一种或几种进行校对,最好用与供试品熔点接近的那一种(见美国药典参比标准品)。依照供试品性质不同,此处列出五种测定熔点或熔距的方法。如果专论中没有指定采用哪种方法,则用方法Ia测定。混合熔点测定法可作为确证鉴别试验,测定时,将供试固体的熔距与等量的供试品和可信样品的紧密混合物的熔距相比较,如有可能应用相应的美国药典参比标准品。测试中,供试品与混合物的观察值一致,便是化学性质相同的可靠依据。装置I———台标准的熔距测定装置I由一只盛透明热交换液浴的玻璃容器、一根合适的搅拌器、一支精密温度计(见温度计)及可控温的热源组成。根据所需温度选择液浴,一般采用液态石蜡油,对高温范围而言则适合采用液态硅油。加入的热交换液应足以浸没温度计分浸线,温度计汞球距容器底部约2cm。可以用明火或电加热,毛细管约长l0cm,内径为0.8—1.2mm,壁厚0.2~0.3mm。装置Ⅱ——可以在方法1, 1a和1b中使用一个仪器。装置Ⅱ比较适宜的熔距测量例子是由一块可以控制加热速率的金属块,和一个监测其温度的传感器组成。此金属块可容纳含有供试品的毛细管并可以监测熔化过程,典型的是依靠一束光和一个检测器。此监测器的信号可能由微电脑处理来测定和显示熔点或熔距,或检测器信号可能绘制出熔点或熔距的可目视估计的曲线。操作步骤(方法l装置I)将供试品研成很细的粉末。除另有规定外,样品若含结晶水,则按专论规定的温度进行干燥脱水:若不含结晶水,则在合适的干燥剂上干燥16小时以上。取一根玻璃毛细管,一端熔封,将足够量的干燥的供试品粉末装入其中,轻轻地在坚实的表面上叩击,使粉末尽可能紧密地集结在管底,形成高2.5—3.5mm的柱体。将热交换液浴加熟,当温度上升至比预期熔点约低30℃时,取出温度计,以一滴热交换液润湿或其他方法,迅速将毛细管贴在温度计上,并调节其高度,使毛细管内的粉末柱与温度计汞球处于同一水平位置。将温度计再插入热交换液浴,在恒定搅拌下继续加热,升温速率约每分钟3℃。当温度上升至比预期熔距下限低约3℃时,降低升温速率至每分钟1~2℃,继续加热,直至供试品全熔。在测定过程中,观察毛细管内供试品柱,把管壁上任何一点确实塌陷时的温度定义为“初熔”,供试品全部液化时的温度定义为“终熔”或熔点。此两点的温度都应在熔距限内。操作步骤(方法Ia,装置I)按方法I,装置I中的规定将制备好的供试品装入毛细管。加热热交换液浴,当温度上升至较预期熔点约低10℃时,控制升温速率每分钟约1±0.5℃。当温度上升至较预期熔距约低5℃时,按方法I,装置I中所述把毛细管插入热交换液浴,继续加热,直至供试品全熔为止。按方法I,装置I的规定记录熔距。操作步骤(方法Ib,装置I)将供试品至于密闭容器内,冷却到10℃或更低,保持至少2小时。在没有前文研细的情况下,将冷却后的物料按方法I,装置I中的规定装入毛细管中,然后立即将充填后的毛细管至于真空干燥器内并在小于20mm汞柱的压力下干燥3小时。然后快速从上放打开干燥器,熔封毛细管的底端,尽可能快地按下面的方法测定熔距:将热交换液加热至预期熔距下限以下10±1℃,然后将毛细管插入热交换液,并以每分钟3±0.5℃的速率升温,直到全熔。按方法I,装置I中的规定记录熔距。操作步骤(方法I,装置Ⅱ)按方法I,装置I中的规定制备供试品并装填如毛细管中。按制造商的操作指南操作仪器装置。加热金属块,直到低于预期熔点约30℃。往加热中的金属块中插入毛细管,并持续以每分钟1~2℃的速率升温直至全熔。检测器信号第一次显示的初始值定义为初熔,检测器信号达到的最终值定义为终熔,或熔点。这两个温度都应在熔距的限度之内。若有争议,只有按方法I,装置I中的规定得到的熔距或温度是正确的。操作步骤(方法Ⅱ)小心地将供试品在尽可能低的温度下熔化,将其移入2端都开口的毛细管中,填入大约10mm。将填充后的毛细管冷却到10℃或更低,保持至少24小时或用冰直接接触至少2小时。然后将毛细管用适当的方法贴在精密温度计上,在水浴中调节毛细管位置,使物料的上边缘在水平面下10mm,按方法I,装置I中的规定加热,但是,当达到预期熔解点5℃以下时,调节升温速率至每分钟0.5~1.0℃。观测到物料在毛细管中上升的那个温度,就是熔点。操作步骤(方法Ⅲ)将定量的供试品缓慢熔化,在达到90~92℃之前的一段时间内搅拌。移去热源并将熔化了的物质冷却到高于预期熔点的8~10℃。冷却合适的温度计的汞球处(见温度计(21))至5℃,擦干,当它仍然冷的时候,将汞球浸入熔融的物质中直到下方半个汞球被浸没。立即移开汞球,远离热源,保持温度计垂直于水平面,直至腊状物表面变的无光泽,将其浸入不高于16℃的水浴5分钟。将温度计安全的固定在试管中,使最低点高于试管底部15mm。将试管悬挂于水浴中,调节到16℃,以每分钟2℃的升温速率将水浴升至30℃,然后改变审问速率为每分钟1℃,并记录第一滴熔融物质滴落温度计的温度。用新近熔化的供试品再重复测定2次。如果3次测定的偏差小于1℃,将三个温度的平均值作为熔点。如果三次测定的偏差大于等于1℃,再测定2次,取5次测试的平均值为熔点。
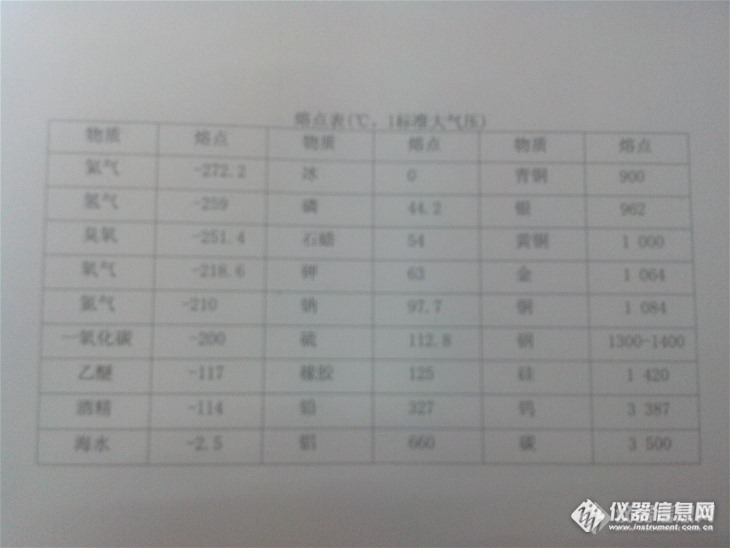
物质熔点测定方法物质的熔点,一般情况下就是指该物质固、液两相可以共存并处于平衡的温度,一说到这个,我们就会想到冰和水,以冰熔化成水为例,在一个大气压下冰的熔点是0℃,而温度为0℃时,冰和水可以共存在通常的情况下,物体在不同的温度下,其状态也是不同的,且即时在同一温度,其状态也是不确定的,如晶体处于熔点温度时,可能有三种状态,即可能固态、可能液态、也可能固液共存,常用物质溶解表如下:熔点表http://ng1.17img.cn/bbsfiles/images/2013/11/201311131143_476934_2154459_3.jpg那么上表这些物质的熔点怎么得来的的呢?有没有什么方法可以测试物质的熔点呢,那就看看吧!1.原理以加热的方式使毛细管中的试样从低于其初熔时的温度逐渐升至高于其终熔时的温度,通过目测分别观察其初熔和终熔温度,即为试样的初熔点和终熔点。2.仪器(1)毛细管:硬质玻璃毛细管,内径0.9~1.1mm,壁厚0.15~0.2mm,长110~120mm,一端封口。(2)测量温度计:分度值为0.1℃,长约400mm,全浸或局浸式并经校正的温度计,根据熔点范围选择适当量程。(3)辅助温度计:分度值为1℃的普通温度计,量程与测量温度计一致。(4)加热器:加热均匀、安全、易控制温度的加热装置。(5)熔点测定装置:容积250ml的内热式烧瓶,直径80mm,瓶长130~140mm,瓶口处有一温度计插口直径约10mm的圆孔,在瓶口上方有两个相对称的毛细管插口直径约2mm的圆孔。3.传热液体201型甲基硅油(粘度在500号以上),或硫酸与硫酸钾混合液(220ml浓硫酸中加入70~80g无水硫酸钾,再加入0.1~0.2g硝酸钾)。4.样品的干燥取少量混匀、研细的试样放于干燥、清洁的表面皿中,于105℃干燥箱中干燥30min后取出,放在干燥器中冷却备用。5.测定将少量干燥、研细的试样装入清洁、干燥、一端封口的毛细管中,取一高约800mm、直径10mm的干燥玻璃管直立于玻璃板上,将装有试样的毛细管经玻璃管投掷8~10次,直至毛细管内试样紧缩至3-4mm高,再将开口一端封熔,圆底烧瓶中注入其体积四分之三的传热液体,测量温度计插入试管内的传热液体中,并使温度计的中间泡也浸没于传热液体中,温度计不得碰及管底。加热使传热液体温度缓缓上升至熔点前10℃时,将装有试样的毛细管通过锥形导管插入试管内并附着于温度计水银球的中部,继续加热,调节加热器使温度上升速度为0.5~1.0℃/min。观察毛细管中试样熔化情况,当试样开始出现明显的局部液化现象时的温度为初熔点。6.结果的表示与计算若测定中使用的是局浸式温度计,初熔点t=t+△t;若测定中使用的是全浸式温度计,初熔点t按下式计算: t=t1+△t1+△t2+△t3△t1=0.00016h1(t1-t2)△t2=0.00016h2(t1-t3)式中 t——初熔点,℃△t1——温度计露出液面至塞口处的水银柱校正值,℃△t2——温度计露出塞外的水银柱校正值,℃△t3——温度计的示值校正值,℃t1——观察温度,t2——试管内液面至塞口的中间处的温度,t3[
要做实验测定!1、熔点的测定化合物的熔点是指在常压下该物质的固—液两相达到平衡时的温度。但通常把晶体物质受热后由固态转化为液态时的温度作为该化合物的熔点。纯净的固体有机化合物一般都有固定的熔点。在一定的外压下,固液两态之间的变化是非常敏锐的,自初熔至全熔(称为熔程)温度不超过0.5-1℃。若混有杂质则熔点有明确变化,不但熔点距扩大,而且熔点也往往下降。因此,熔点是晶体化合物纯度的重要指标。有机化合物熔点一般不超过350℃,较易测定,故可借测定熔点来鉴别未知有机物和判断有机物的纯度。在鉴定某未知物时,如测得其熔点和某已知物的熔点相同或相近时,不能认为它们为同一物质。还需把它们混合,测该混合物的熔点,若熔点仍不变,才能认为它们为同一物质。若混合物熔点降低,熔程增大,则说明它们属于不同的物质。故此种混合熔点试验,是检验两种熔点相同或相近的有机物是否为同一物质的最简便方法。熔点装置图:2、沸点的测定液体的分子由于分子运动有从表面逸出的倾向,这种倾向随着温度的升高而增大,进而在液面上部形成蒸气。当分子由液体逸出的速度与分子由蒸气中回到液体中的速度相等,液面上的蒸气达到饱和,称为饱和蒸气。它对液面所施加的压力称为饱和蒸气压。实验证明,液体的蒸气压只与温度有关。即液体在一定温度下具有一定的蒸气压。当液体的蒸气压增大到与外界施于液面的总压力(通常是大气压力)相等时,就有大量气泡从液体内部逸出,即液体沸腾。这时的温度称为液体的沸点。通常所说的沸点是指在101.3kPa下液体沸腾时的温度。在一定外压下,纯液体有机化合物都有一定的沸点,而且沸点距也很小(0.5-1℃)。所以测定沸点是鉴定有机化合物和判断物质纯度的依据之一。测定沸点常用的方法有常量法(蒸馏法)和微量法(沸点管法)两种。 实验步骤1、熔点的测定毛细管法:①准备熔点管:将毛细管截成6~8cm长,将一端用酒精灯外焰封口(与外焰成40o角转动加热)。防止将毛细管烧弯、封出疙瘩。②装填样品:取0.1~0.2g预先研细并烘干的样品,堆积于干净的表面皿上,将熔点管开口一端插入样品堆中,反复数次,就有少量样品进入熔点管中。然后将熔点管在垂直的约40cm的玻璃管中自有下落,使样品紧密堆积在熔点管的下端,反复多次,直到样品高约2~3cm为止,每种样品装2~3根。③仪器装置:将b形管固定于铁架台上,倒入液体石蜡做为浴液,其用量以略高于b形管的上侧管为宜。将装有样品的熔点管用橡皮圈固定于温度计的下端,使熔点管装样品的部分位于水银球的中部。然后将此带有熔点管的温度计,通过有缺口的软木塞小心插入b形管中,使之与管同轴,并使温度计的水银球位于b形管两支管的中间。④熔点测定:粗测:慢慢加热b形管的支管连接处,使温度每分钟上升约5℃。观察并记录样品开始熔化时的温度,此为样品的粗测熔点,作为精测的参考。精测:待浴液温度下降到30℃左右时,将温度计取出,换另一根熔点管,进行精测。开始升温可稍快,当温度升至离粗测熔点约10℃时,控制火焰使每分钟升温不超过1℃。当熔点管中的样品开始塌落,湿润,出现小液滴时,表明样品开始溶化,记录此时温度即样品的始熔温度。继续加热,至固体全部消失变为透明液体时再记录温度,此即样品的全熔温度。样品的熔点表示为:t始熔~t全熔。实测:尿素(已知物,133~135℃)、桂皮酸(未知物,132~133℃),混合物(尿素-桂皮酸=1:1,100℃左右)。实验过程中,粗测一次,精测两次。2、沸点的测定微量法测定沸点:①沸点管的制备:沸点管由外管和内管组成,外管用长7~8厘米、内径0.2~0.3cm的玻璃管将一端烧熔封口制得,内管用市购的毛细管截取3~4cm封其一端而成。测量时将内管开口向下插入外管中。②沸点的测定:取1~2滴待测样品滴入沸点管的外管中(思考题9),将内管插入外管中,然后用小橡皮圈把沸点附于温度计旁,再把该温度计的水银球位于b形管两支管中间,然后加热。加热时由于气体膨胀,内管中会有小气泡缓缓逸出,当温度升到比沸点稍高时,管内会有一连串的小气泡快速逸出。这时停止加热,使溶液自行冷却,气泡逸出的速度即渐渐减慢。在最后一气泡不再冒出并要缩回内管的瞬间记录温度,此时的温度即为该液体的沸点,待温度下降15~20℃后,可重新加热再测一次(2次所得温度数值不得相差1℃)。按上述方法进行如下测定:CCl4沸点(76℃)。 注意事项1.熔点管必须洁净。如含有灰尘等,能产生4—10OC的误差。2.熔点管底未封好会产生漏管。3.样品粉碎要细,填装要实,否则产生空隙,不易传热,造成熔程变大。4.样品不干燥或含有杂质,会使熔点偏低,熔程变大。5.样品量太少不便观察,而且熔点偏低;太多会造成熔程变大,熔点偏高。6.升温速度应慢,让热传导有充分的时间。升温速度过快,熔点偏高。7.熔点管壁太厚,热传导时间长,会产生熔点偏高。
实验室有一台数显熔点仪,但有时样品测定的结果有偏差,特别是和供应商提供的检测报告误差有2度,有时在质量指标的边缘,很难判断。所以想购买一个老式的油浴装置,温度计已经买了,但市场上很难看到烧杯配加热管的装置,各位谁有看到,推荐一下,最好在南京市内。
求JB/T 20187-2017 溶出度测定装置
熔点仪的特点:主要用于染料、药物、香料等晶体有机化合物熔点之测定,以便确定其纯度。丈量方法完全符合药典标准,一般最多可同时丈量三根样品,自动计算初、终熔均匀值。分为目视熔点仪,数字熔点仪,微机熔点仪,显微熔点仪等几大类。 熔点仪原理,根据物理化学的定义,物质的熔点是指该该仔细搜索由固态变成液态时的温度。在有机化学领域中,熔点测定是辨认物质本性的基本手段,也是纯度测定的重要方法之一。因此,熔点测定仪在化学产业、医药研究中占有重要地位,是生产药物、香料、柒料及其他有机晶体物质的必备仪器。 测定方法:测定熔点方法一般用毛细管法和微量熔点测定法,在实际操纵中,我们就用专业的熔点测定仪来测物质熔点。通过熔点仪,我们可以测定样品的熔点值。通过和纯物质的熔点值对比就可以得到样品的纯度情况。因此,熔点测定仪在化学产业、医药研究中占有重要地位,是生产药物、香料、染料及其他有机晶体物质的必备仪器。应用:熔点仪在化学产业、医药研究中具有重要地位,是生产药物、香料、染料及其他有机晶体物质的必备仪器。
国标17657-2013 4.59甲醛释放含量-干燥器法,温度测定装置描述是:温度测定装置,例如热电偶,温度测量误差0.1℃,放入干燥器中,并把干燥器紧邻其他放有试件的干燥器。请问这温度测定装置只用于测空白干燥器的温度吗?有哪位大神对这方法了解的,可否讲解一二呢,非常感谢了
残炭是在残炭测定装置的坩埚中,将试油按规定的条件,加热到试油蒸发分解而形成的焦黑色残留物。电炉法规定炉温保持520±5℃下蒸发分解后的残留物。一般柴油残炭规定把试油蒸馏到残余10%后,才蒸发分解。称10%蒸馏残余物残炭,这种10%蒸馏残炭物残炭值比全烧残炭结果大得多,重质燃料油规定做全残炭。 大型低速柴油机可使用含残炭10%的重质燃料油。残炭值影响燃烧室的结焦结炭。但对气缸和活塞的磨损则不仅取决于残炭的多少,还主要取决于炭质的软硬。含硫高的积炭坚而硬,磨损较大。残炭检测优选A1260微量残炭测定仪。[font=&]这是得利特(北京)公司的油品分析仪器,性能挺稳定的。适用于化工、电力、石油等行业。得利特主要我公司产品有:闪点测定仪 ,运动粘度测定仪,微量水分测定仪,油液污染度检测仪、酸值测定仪、微量水分测定仪、凝点倾点测定仪、体积电阻率测定仪、微量残碳测定仪等多种润滑油分析仪器、燃料油分析仪器、绝缘油分析仪器,水质分析检测仪器、气体检测仪器。[/font]
【作者】: 【题名】:总氮测定装置及总氮总磷测定装置【期刊】:【年、卷、期、起止页码】:【全文链接】:https://t.cnki.net/kcms/detail?v=kxaUMs6x7-4I2jr5WTdXti3zQ9F92xu093x413AcgcHHtMVf2jH7MavFQh-Y9kig2GYNk3M9pnH-HH_iKcKa0N4RKWi1lFcy&uniplatform=NZKPT
水洗筛余物测定装置GY-60型安装和使用指导[font=&][size=13px][color=#333333]水洗筛余物测定装置GY-60型整机安装并调试合格后出厂,用户只需接上水管和电源就可以直接使用,操作十分方便,是测定炭黑、白炭黑以及其它粉粒物质筛余物不可缺少的装置,比美国ASTMD1514-80标准推荐使用的装置体积少四分之三。[/color][/size][/font]
请问显微熔点测定仪上面的上限温度和下限温度是什么意思?具体该怎么设置?
测定的结果都是熔程,看标准说"平行测定5次,舍去最大最小值,求剩余3个数据的平均值为物质的熔点”,最大最小值舍去好说,这个“平均值”是怎么求取的呢?难道是:3个初熔、终熔各求平均值?如果是这样,所得的结果有依据吗?依据是什么?想不明白,但求各位,不胜感激!!
[color=#ff0000] [b]水洗筛余物测定装置安装和使用:[/b][/color] 水洗筛余物测定装置整机安装并调试合格后出厂,用户只需接上水管和电源就可以直接使用,操作十分方便,是测定炭黑、白炭黑以及其它粉粒物质筛余物不可缺少的装置,比美国ASTMD1514-80标准推荐使用的装置体积少四分之三。
有没有能够满足铁合金中C%0.1%的碳硫分析仪,除红外吸收外.用高频燃烧装置能配自动吸收装置.
咨询一下,大家都用什么牌子的显微熔点测定仪,国产的,性价比高一些的,谢谢!
[align=left] 苏州市计量测试院理化检测室新建的渗透压摩尔浓度测定仪检定装置顺利通过江苏省质量技术监督局考核。该计量标准能够开展测量范围为(0~700)mOsmol/kg的渗透压摩尔浓度测定仪的检定、校准工作。截止到11月底,已为苏州及周边地区10余家医药企业的20余台仪器提供了检校服务,满足了相关仪器的溯源需求。[/align][align=left] 渗透压摩尔浓度测定仪采用冰点下降原理,间接测定溶液的渗透压摩尔浓度,它广泛应用在生物医药行业。人体体液需要保证一定范围的渗透压,在制备注射剂、眼用液体制剂等药物制剂时,必须保证其渗透压;添加了渗透压调节剂的制剂,也应控制其渗透压摩尔浓度。此类仪器的测量精度和稳定性是保证医药产品渗透压准确可靠的必要前提。[/align]
请教有关化学样品DSC熔点测定与毛细管熔点测定的区别

药物熔点的测定 熔点是物质的物理常数。测定熔点可以鉴别药物,也可以反映药物的纯杂程度。1. 实验原理熔点的定义:固液两相的蒸气压相同而且等于外界大气压时的温度就是该固体物质的熔点。2. 实验仪器及药品仪器:数字熔点测定仪MP430,研钵等;药品及实际:原料药,蒽,糖,柠檬酸(样品均为客户送检),乙醇,去离子水。3. 实验步骤1)样品填装:研碎,填装,2~3mm为宜(一般是指熔化后的样品高度),装填时依据样品的形状不同装填合适的高度。http://ng1.17img.cn/bbsfiles/images/2013/07/201307310936_454871_2599013_3.jpg2)开机测试:将装填好的毛细管放入仪器中,设置好参数,升温开始测试,选择手动测试,目视观察样品的熔化过程,确定初熔和终熔点。http://ng1.17img.cn/bbsfiles/images/2013/07/201307310937_454874_2599013_3.jpg测试完毕后,保存实验数据并打印。http://ng1.17img.cn/bbsfiles/images/2013/07/201307310937_454873_2599013_3.jpg4. 实验结果样品编号样品名称初熔(℃)终熔(℃)1原料药273.9274.5273.7274.42恩215.2216.1215.2216.33糖150.3151.5150.3151.74柠檬酸149.6[align=center
需要测试渗透探伤剂中汞,砷,锌,铅等低熔点金属含量测定,其中汞铅含量小于1ppm。是否能做?据国内的供应商说不能做,但是我们有美国供应商做过相关实验的报告。
[em17] 请问我们实验室新来一台显微熔点测定仪,但是我不怎么会用,哪位大侠能把你的心得弄上来给我看看啊?我这里感激流涕啊
有没有粗略测定高分子熔点的方法?我知道一个双浴式熔点测定的方法,设备挺简单,不知道是否可行?
哪位大哥有MANOMATRIC压差式BOD测定装置的说明书,小弟不胜感谢!
测定物质熔点时,要求研磨样品但是这个样品的细度?多少目?要具体的要求不 ?细度不到,对所测的熔点的值有多大的影响呢
熔点是指物质在大气压力下固态与液态处于平衡时的温度。固体物质熔点的测定通常是将晶体物质加热到一定温度时,晶体就开始由固态转变为液态,测定此时的温度就是该晶体物质的熔点。熔点测定是辨认物质本性的基本手段,也是纯度测定的重要方法之一。因此,熔点仪在化学工业、医药研究中据有重要地位,是生产药物、香料、染料及其他有机晶体物质的必备仪器,也是实验室常用的基础仪器之一。纯净的固体有机物,一般都有固定的熔点,而且熔点范围(又称熔程或熔距,是指由始熔至全熔的温度间隔)很小,一般不超过0.5—1℃;若物质不纯时,熔点就会下降,且熔点范围就会扩大。利用这一性质来判断物质的纯度和鉴别未知化合物。例如,一个未知化合物,测得其熔点与某一已知化合物的熔点相同或者十分相近时,将未知样品与已知样品等量混合后测定其混合熔点。若熔点没有变化,且熔点范围不超过1℃时,一般可以认为二者是同一物质,如果混合熔点发生变化,熔点范围大,则可判定它们不是同一物质。这种鉴定方法叫做混合熔点法。测量熔点的方法有两种:一种是毛细管熔点测定法,另一种则是熔点仪测定熔点法。基于科技的发展与进步,熔点仪不断的更新换代,实现了许多实用性功能,且操作方便,数据精确。因此实验室常用的是采用熔点仪测定熔点的方法。目前全球使用最广泛的熔点仪是美国斯坦佛大学研究所最新研究成果的MPA100全自动熔点仪。MPA100熔点仪是按照药典规定的熔点检测方法而设计的,MPA100熔点仪利用电子技术实现温度程控,初熔和终熔数字显示。应用了线性校正的铂电阻作为检测元件,并用电子线路实现了快速“起始温度”设定,三通道同时设定及可供选择的线性的升温速率。MPA100熔点仪采用药典规定的毛细管作为样品管,通过高分辨率的数码成像检测器观察毛细管内样品的熔化过程,清晰直观,是制药、化工、燃料、香料、橡胶等行业理想的熔点检测仪器。http://www.sinoinstrument.com/UploadFiles/Image/s2013050910524395610(2).jpg
我们新做个一个产品,熔点是295~299度,原来的毛细管熔点仪不能测定,请问那一种熔点仪可以测定,谢谢!
[url=http://www.f-lab.cn/stereotaxis/a-2.html]NARISHIGE的动物肌肉固定装置A-2[/url]通过抓握肌肉或神经纤维的两端,并且轻轻地拉伸两端保持张力,从而促进微电极插入肌肉或神经纤维。需要注意的是NARISHIGE的动物肌肉固定装置A-2是NARISHIGE的A-3的一个组成部分,使用时需要A-3的其他组件。如果要单独使用动物肌肉固定装置A-2,我们建议使用NARISHIGE的X-阻止装置将其连接到平台。[img=动物肌肉固定装置]http://www.f-lab.cn/Upload/a-2_.jpg[/img]动物肌肉固定装置:[url]http://www.f-lab.cn/stereotaxis/a-2.html[/url]
请问熔点仪、显微熔点仪、药物熔点仪,有何区别?一直搞不清楚。多谢先
[em09502]一、简述测量依据:JJG307-1988《交流电度表 三相三有功电度表DS862-2 DS862-4 检定规程》。测量标准:三相电能表 导轨式安装电能表ADL300-EF/C 检定装置,型号SDX-1,规格3×(100~400)V;3×(0.1~50)A,准确度级别0.2级。环境条件:温度(20±2)℃,相对湿度(35~85)%。测量对象:三相四线有功电能表,准确度等级2.0级,型号DT241X-4,规格3×380/220V;3×1?郾5(6)A。测量过程:装置输出一定功率给被检表,并对被检表进行采样积分,得到的电能值与装置输出的标准电能值比较,得到被检表在该功率时的相对误差。二、数学模型 r="r0" 式中:r———被检电能表的相对误差;r0———三相电能表检定装置上测得的相对误差。三、输入量的标准不确定度评定输入量r0的标准不确定度u(r0)的来源主要有两个方面:在重复性条件下,对被测电能表测量其典型测量点引起的不确定度分量u(r01),采用A类评定方法;由三相电能表检定装置的误差引起的不确定度分量u(r02)采用B类评定方法。标准不确定度分量u(r01)的评定通过对2.0级被测电能表在3×380/220V;3×1.5A;cosΦ=1.0的量程上重复测量了10次,每一次测量都启动控制按键,测得结果如表1所示。标准不确定度分量u(r)=S(r)=0.028% 标准不确定度量分量u(r)的评定该不确定度分量主要由本相电能表检定装置的误差引起。它包括:三相标准功率电能表的不确定度u=0.2%/;三相标准功率电能表的数字显示分辨率带来的不确定度u=0.29×0.01%;误差数据化整间隔带来的不确定度u3=0.29×0.2%;标准电流互感器 电流互感器LDZ1 引起的不确定度u4≤0.02%;数控光电采样器带来的不确定度u5≤0.02%。标准不确定度分量u(r02)=0.1323% 输入量r的标准不确定度u(r)的计算 u(r)=[u2(r)+u2(r)]1/2=[0.0282+0.13232]1/2%=0.14%四、合成标准不确定度的评定合成标准不确定度汇总于表2。五、扩展不确定度的评定取置信概率p=95%、包含因子k=2,则扩展不确定度 U95=kuc(r)=2×0.14%=0.28% 六、测量不确定度报告本装置对2.0级三相线有功电能表在3×380/220V;3×1.5A;cosΦ=1.0时,相对误差测量结果的扩展不确定度U95=0.28% 测量不确定度验证按照《计量标准考核规范》规定的验证方法,采用其中传递比较法进行验证。将被测2.0级的三相四线有功电能表用本装置测量后,再送省计量院,用高两个级别的三相电能表检定装置(准确度级别为0.05级、测量时的扩展不确定度U0=0.036%)测量,测量的结果如表3所示。验证公式为:|y-y|≤;由于U≤成立, 故公式为|y-y0|≤U95 验证结果最大差值为:|0.44-0.63|≤0.28%、即0.19%≤0.28%, 证明:测量准确、可靠。
熔点仪的操作步骤,常规熔点测定: 1.开启电源开关,稳定20分钟,此时,保温灯、初熔灯亮、电表偏向右方,初始温度为50℃左右。 2.通过拨盘设定起始温度,通过起始温度按钮,输入此温度,此时预置灯亮。 3.选择升温速率,将波段开关调至需要位置。 4.预置灯熄灭时,起始温度设定完毕,可插入样品毛细管。此时电表基本指零,初熔灯熄灭。 5.调零,使电表完全指零。 6.按下升温钮,升温指示灯亮(注意!忘记插入带有样品的毛细管按升温钮,读数屏将出现随机数提示您纠正操作)。 7.数分钟后,初熔灯先闪亮,然后出现终熔读数显示,欲知初熔读数,按初熔钮即得。 8.只要电源未切断,上述读数值将一直保留至测下一个样品。 连接熔点仪和计算机 1.将随机所附的光盘插入计算机。 2.计算机进入系统执行WRS程序,可实现测试过程中熔化曲线的绘制,结果的显示。
GBT 24892-2010 动植物油脂 在开口毛细管中熔点(滑点)的测定







 我要推广仪器
我要推广仪器
 下载APP
下载APP