各位大佬,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]内标法,做内标标准曲线,是将等量的内标物加入到等体积不同浓度的标准物质中去,然后进色谱检验吗?
请问许多文献上提出核磁内标法(绝对测量法)定量分析时不需引进任何校正因子,不需制作标准曲线,但高效液相色谱为何需制作标准曲线?高效液相色谱和核磁内标法定量的原理差不多呀?[em09]
[color=#444444]请问,液相色谱测定含量双样双针的,f值有没有统一规定的啊?F=称样量/A峰面积/稀释倍数,要不要除以对照品的稀释体积啊?是随便写还是有统一规定写法啊?[/color]
灭多威原药的分析方法一:灭多威的其他名称、结构式和基本物化参数如下:ISO通用名称:MethomylCIPAC数字代码;264化学名称:1-(甲硫基)亚乙基氨甲基氨基甲酸酯结构式 :CH3NH-CO-O-N=C-SCH3 CH3经验式:C5H10N2O2S相对分子质量:162.2(按1989年国际相对原子质量计)生物性质:内吸性杀虫剂熔点:78-79oC蒸汽压:(25oC):666.6X10-5Pa溶解度:(g /L25oC):水58 g /L,甲醇1000 g /L,丙酮730 g /L,乙醇420 g /L。稳定性:固体状态稳定,中性和微酸性介质中稳定,碱性溶液中缓慢分解,潮湿土镶中易分解。二:方法概要灭多威含量采用高效液相色谱法测定,UV检测器,254nm波长,使用乙酰苯胺内标物,内标面积法定量。三:鉴别试验:液相色谱法,在相同的色谱操作条件下,试样溶液主峰的保留时间与标样溶液色谱峰的保留时间,其相对偏差在1.5%以内,灭多威原药进行反相液相色谱测定.。四:仪器高效液相色谱仪:UV检测器,高压输液泵色谱柱:C18高效液相反相色谱柱过滤器:滤膜孔径:0.45nm进样针:50ul色谱工作站:高效液相色谱工作站五:试剂乙腈:色谱纯蒸馏水:双重蒸馏水,0.45um水膜过滤。灭多威标样:纯度:99.0%(m/m)内标溶液:精确称取5g乙酰苯胺内标物,放入干净的烧杯中,加入95g乙二醇,加热溶解,冷却备用。六:色谱操作条件流动相:乙腈/水=100/400,每500ml加入5滴磷酸调PH值,超声30min备用。稀释液:乙腈/水=100/400,超声30min备用。波长:254nm流速:1.00ml/min[size=11.0p
液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的外标法和内标定量有何区别?是如何处理的?
有用过戴安双三元液相色谱的吗?其在线固相萃取的重现性好不
问题: 请教一个问题,赛默飞的双三元液相色谱是超高效液相色谱吗?回复: 就是普通的液相 ,有两个泵,可以当两台仪器用,可以分离维生素,和实现苯并芘的在线分析。
有内标物时高效液相色谱法中如何计算含量
GB/T 23383-2009 食品中双乙酸钠的测定 高效液相色谱法 2009年5月1日实施。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152564]GB/T 23383-2009 食品中双乙酸钠的测定 高效液相色谱法[/url]
昨天参加了戴安液相色谱和离子色谱CDC座谈会,听了他们刘经理讲解了下戴安双三元液相色谱在液相色谱分析中的优势,特别是其在线固相萃取的应用,我觉得真的可以大大减轻我们检验人员的工作量,大大的缩短了我们的检测时间哦。请问有朋友用过戴安双三元的液相色谱吗?我要是说 话能算数的话我肯定马上就开始计划购买哦
随着分析方法的发展及科学研究的不断深入,复杂体系分离已成为分析化学研究的热点和难点之一。双三元液相色谱仪将两个液相泵集于一体,结合独特的阀切换技术及流路连接设计,可以覆盖常规分析和复杂样品的二维分析等
戴安双三元液相色谱如何性价比如何,价格多少?
硫双威原药该产品有效成分硫双威的结构式和基本物化参数如下:化学名称:3,7,9,13-四甲基-5,11-二氧杂-2,8,14-三噻-4,7,9,12-四-氮杂十五烷-3,12-二烯-6,10-二酮结构式http://ng1.17img.cn/bbsfiles/images/2014/12/201412271243_529409_2960432_3.png 实验式:C10H18N4O4S3相对分子质量:354.5(按2007年国际相对原子质量计)生物活性:杀虫熔点:(173~174)℃蒸气压(20℃):5.1Pa溶解度:水中35mg/L,丙酮中8g/kg,甲醇中5g/kg,二甲苯中3g/kg。稳定性:60℃稳定,其水悬浮液因日光而分解,pH6稳定,pH9迅速水解,pH3缓慢水解。 1 范围本标准适用于硫双威及其生产中产生的杂质组成的硫双威原药。2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 1600-2001 农药水分的测定方法GB/T 1601-1993 农药pH值的测定方法GB/T 1604-1995 商品农药验收规则GB/T 1605-2001 商品农药采样方法GB 3796-2006 农药包装规则GB/T 6682-2008 分析实验室用水规格和试验方法GB/T 19138-2003 农药丙酮不溶物的测定方法HG 2611-1994 灭多威原药3 要求3.1 外观:白色至类白色固体。3.2硫双威原药应符合表1要求。表1 硫双威原药控制项目指标 项 目指 标硫双威质量分数,% ≥95.0灭多威质量分数,% ≤0.5水分质量分数,% ≤1.0pH值4.0~7.0丙酮不溶物质量分数,% ≤0.54 试验方法除另有说明,本试验所使用的试剂均为分析纯试剂,水应符合GB/T6682-2008中的三级水规格。4.1 抽样按照GB/T1605-2001中“商品原药采样”进行,用随机法确定抽样的包装件;最终抽样量不少于100g。4.2 鉴别试验 本鉴别试验可与硫双威含量的测定同时进行。在相同的色谱操作条件下,试样中某一色谱峰的保留时间与标样溶液中硫双威色谱峰的保留时间,其相对差值应在1.5%以内。4.3 硫双威质量分数的测定4.3.1方法提要试样用50%(V/V)四氢呋喃甲醇溶液溶解,以甲醇+水为流动相,使用Kroasil C18为填充物的不锈钢柱和可调波长紫外检测器,对试样中的硫双威进行高效液相色谱分离和测定。4.3.2试剂和溶液 水:新蒸二次蒸馏水,经0.45μm滤膜过滤;甲醇:色谱纯;四氢呋喃:HPLC; 硫双威标样:已知含量,≥99.0%。4.3.3仪器高效液相色谱仪:具可变波长紫外检测器;色谱数据处理机;色谱桩:250mm×4.6mm(id)不锈钢柱,内填Kroasil C18[
液相色谱测TBBPA(四溴双酚a),峰的基线不平是为什么?
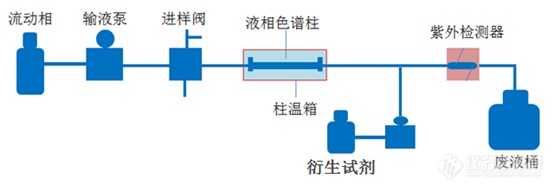
[align=center][size=21px][/size][/align][align=center][font=宋体]液相色谱衍生化法[/font][/align][font=宋体] 衍生化是一种利用化学变换把化合物类似化学结构或具有某种特性的物质的方法。通过衍生化法可以使样品中不易分离或分析的物质易于分离、分析。衍生化过程是使样品与衍生试剂在特定的设备内均匀混合,并在特定的条件下(如特定的温度、压力、光照等)充分反应。[/font][font=宋体] 在色谱分析中衍生化法一般有前处理衍生、柱前衍生、柱中衍生、柱后衍生等,液相色谱中柱后衍生应用较多,被称为柱后衍生液相色谱分析法。[/font][align=center][img=,555,191]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090917333555_9481_2369266_3.png!w555x191.jpg[/img][/align][img]file:///C:/Users/Administrator/AppData/Local/Temp/msohtmlclip1/01/clip_image002.jpg[/img][font=宋体] 衍生化常用的反应有酯化反应、酰化反应、硅烷化反应、环化反应、紫外衍生化反应、荧光衍生化反应等,通过给被测物加入衍生化试剂,再在加热等特定条件下使其发生衍生化反应。比如甲醛直接用液相色谱检测效果不好,而甲醛与[/font]2,4-[font=宋体]二硝基苯肼反应生成红色的[/font] 2,4-[font=宋体]二硝基苯腙,[/font]2,4-[font=宋体]二硝基苯腙用液相色谱检测效果就好的多。呋喃丹、甲萘威等农药成分用液相色谱检测效果不好,衍生化后用高效液相色谱荧光检测器检测效果很好。[/font][font=宋体] 像黄曲霉菌类,如黄曲霉毒素[/font]B1[font=宋体]在紫外光照射下可发出特定荧光,该类物质可以用特定的紫外光照射衍生化荧光检测器检测。[/font][font=宋体] 试剂衍生有的物质需要一种试剂一步衍生,有的物质需要多种试剂多布衍生,那衍生设备也就有单泵单衍生器和多泵多衍生器(通常是双泵双衍生器)不同配置,不管是单泵单衍生器还是多泵多衍生器现在大家基本都集成到一台设备里了。[/font][font=宋体] 美国[/font]Pickering[font=宋体]柱后衍生仪在国内占了很大一部分市场,国内柱后衍生仪主要厂家有天津奥特赛恩斯仪器有限公司、苏州华美辰、北京创新通恒等。[/font][align=center][img=,690,334]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090917557087_1544_2369266_3.png!w690x334.jpg[/img][img=,334,613]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090918005083_2140_2369266_3.png!w334x613.jpg[/img][/align] [font=宋体]随着发展,需要检测的物质会越来越多,液相色谱衍生化法应用的也会越来越多,衍生化法还会有较大发展空间。[/font][size=16px][/size]
【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,0.025 mol/L磷酸溶液-乙腈(87:13)用三乙胺调节pH值至3.0为流动相,检测波长为277nm。理论板数按环丙沙星峰计算应不低于2000,环丙沙星峰与内标物质峰的分离度应符合要求,拖尾因子应不大于2.5。内标溶液的制备 取恩诺沙星对照品25 mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,即得。测定法 取乳酸环丙沙星对照品约25 mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀;精密量取该溶液与内标溶液各5ml,置25ml量瓶中,用流动相稀释至刻度,摇匀,取20µ l注入液相色谱仪,记录色谱图;另取本品适量,同法测定。按内标法以峰面积计算,结果与0.7863相乘,即得。
蔬菜、水果及其制品中吡虫啉、多菌灵、甲基硫菌灵、霜霉威、灭多威、霜脲氰残留量的检测方法-超高效液相色谱-质谱/质谱法 唐玉萍1 范围本非标方法规定了蔬菜、水果及其制品中吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰和灭多威残留量的超高效液相色谱-质谱/质谱检测方法。本非标方法适用于蔬菜、水果及其制品中吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰和灭多威残留量的检测,该方法在番茄、番茄酱、梨、脱水洋葱等样品中经过验证。2 规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法3 原理样品用乙酸-甲醇-水溶液提取,C18色谱柱进行分离,用超高效液相色谱-质谱/质谱检测,内标或外标法定量。4 试剂及材料除另有规定外,所用试剂均为分析纯,水为GB/T 6682 规定的一级水。4.1 甲醇:色谱纯。4.2 乙酸铵。4.3 10mmol/L乙酸铵:准确称取1.926g乙酸铵,定容至500mL容量瓶中,配制成50mmol/L乙酸铵。用溶剂过滤装置过0.2µm水相滤膜,0~4℃保存,有效期15天。临用时用水稀释成10mmol/L。4.4 冰乙酸。4.5 甲醇-水溶液(30+70,V/V)。4.6 提取液Ⅰ:乙酸-甲醇-水溶液(0.1+50+50,V/V/V)。4.7 提取液Ⅱ:乙酸-甲醇-水溶液(0.1+80+20,V/V/V)。4.8 吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰、灭多威和D4-吡虫啉标准品:均为德国Dr.公司,纯度≥98.0%。4.9 6种农残标准储备液:分别准确称取吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰、灭多威标准品10mg~15mg(精确到0.1mg)于50mL容量瓶中,用甲醇溶解并定容至刻度,配制成浓度约为200~300µg/mL的标准储备液,于-18℃避光保存,有效期18个月。4.10 中间浓度混合标准溶液:根据需要,取适量6种农残标准储备液,用甲醇-水溶液(4.5)稀释配制成2µg/mL的混合标准中间液,0~4℃保存,有效期6个月。4.11 内标标准储备液:准确称取D4-吡虫啉标准品约10mg(精确到0.1mg)于50mL容量瓶中,用甲醇溶解并定容至刻度,配制成浓度约为200µg/mL的内标标准储备液,于-18℃避光保存,有效期18个月。4.12 中间浓度内标溶液:取D4-吡虫啉标准储备液,用甲醇-水溶液(4.5)逐级稀释配制成4µg /mL和200ng/mL,0~4℃保存,有效期6个月。4.13 混合标准工作溶液:准确吸取一定量的中间浓度混合标准溶液(4.10)和中间浓度内标溶液(200 ng/mL),用甲醇-水溶液(4.5)配制成10,20,50,100,200 ng/mL系列浓度的混合标准工作溶液,内标浓度均为20 ng/mL,0~4℃保存,有效期3个月。4.14 微孔滤膜:0.2µm,有机相。4.15 流动相过滤滤膜:0.2µm,水相。5 仪器和设备5.1 高效液相色谱-串联质谱仪(LC-MS/MS):配有电喷雾离子源(ESI)。5.2 电子分析天平:感量分别为0.1 mg和0.01 g。5.3 超声波水浴。5.4 漩涡混合器:3000r/min。5.5 离心机:9000r/min。5.6 离心管:聚四氟乙烯,50mL。5.7 溶剂过滤装置。6 试样的制备和保存6.1 试样的制备与保存取番茄、梨等果蔬样品约500g,将其可食用部分切碎后,用粉碎机粉碎成浆状,混匀,均分成两份作为试样,分装入洁净的容器中,密封。将试样于-18℃以下冷冻保存。取番茄酱样品约500g,混匀,均分成两份作为试样,分装入洁净的容器中,密封。将试样于-18℃以下冷冻保存。取脱水洋葱样品约200g,混匀,均分成两份作为试样,分装入洁净的容器中,密封。将试样于0~4℃保存。注:在制样过程中,应防止样品受到污染或发生农药残留量的变化。7 测定步骤[
GB/T 30939-2014,化妆品中污染物双酚A的测定 高效液相色谱-串联质谱法,哪位手上有希望分享一下,照片,扫描件都可以,先谢谢了!
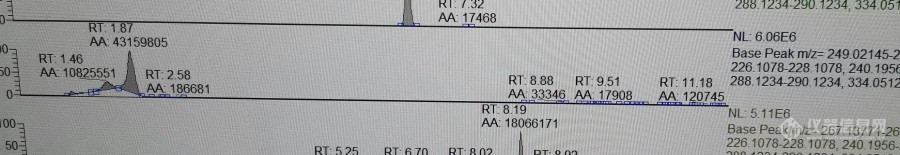
最近在做7种双酚类物质在环境介质中的检测,用到超高效液相色谱仪。目前在建立色谱方法阶段,但是跑标准品发现双酚S的峰型始终改善不好(如下图所示),其余6种(双酚A、F、AF、P、AP、Z)峰型都很好。由于本人是新手,很多东西都不大明白,仪器也不在实验室,因此想向各位大佬请教一下,如果想要改善双酚S的峰型需要从哪个方面入手?希望各位大佬能给出建议~[b]色谱条件[/b]:流动相:A相甲醇,B相水(含2mM甲酸铵,pH用甲酸调到2.98),1-7分钟内甲醇40%-80%梯度提升浓度色谱柱:[font=Arial][font='Noto Sans CJK JP Black',sans-serif]ACQUITY UPLC BEH C18 Column, [/font][font='Noto Sans CJK JP Black',sans-serif]130Å ,1.7 μm, 2.1 mm X 100 mm, 1/pkg[/font][/font][font=Arial][font='Noto Sans CJK JP Black',sans-serif]进样量:5[font=Arial]μL 流速[/font][/font][/font][font=微软雅黑][color=black]:[/color][/font][font=Arial][color=black]0.4ml/min[/color][/font][font=Arial][color=black][/color][/font][font=Arial][color=black]不知道这些条件够不够解决问题......如果需要其他条件还麻烦各位提出来,谢谢~[/color][/font][img=双酚S色谱图,690,118]https://ng1.17img.cn/bbsfiles/images/2020/09/202009081611052239_4364_4229416_3.jpg!w690x118.jpg[/img][img=双酚S结构式,584,214]https://ng1.17img.cn/bbsfiles/images/2020/09/202009081612044342_969_4229416_3.png!w584x214.jpg[/img]双酚S的pKa为8.2

标准全名:SN/T 2282-2009 食品接触材料 高分子材料 食品模拟物中双酚A的测定 高效液相色谱法其中第七点http://ng1.17img.cn/bbsfiles/images/2011/12/201112292136_342540_1627312_3.jpg我估计很少人直接做橄榄油的了,大多都是做油的替代物,一个95%乙醇,一个异辛烷。我觉得95%的乙醇可以直接过045μ滤膜测试,异辛烷需要进一步处理。但是,处理步骤按7.1.2橄榄油模拟浸泡液进行吗?还有,测试乙烯酰胺也一样吧。
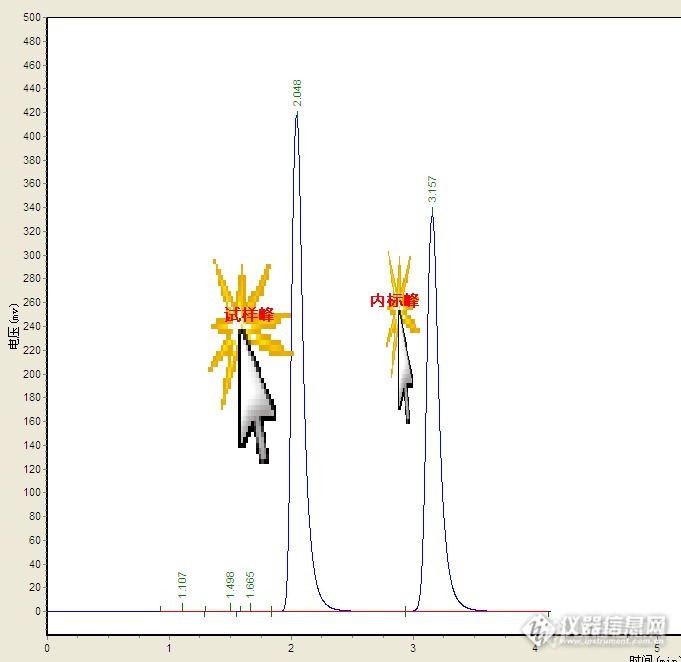
请问一下,做高效液相色谱的内标法,做的是糖的色谱,测得果糖,山梨醇,葡萄糖,蔗糖这四个糖,不知道用哪一种内标物,还有就是使用了内标物之后怎么计算啊?
[b]高效液相色谱法的计算方法[/b]高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。 1.对仪器的一般要求 所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。 在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。 正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变, 以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。 2.系统适用性试验 按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子. (1) 色谱柱的理论板数(n) 在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。 (2) 分离度 定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。 (3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。 3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。 (2) 主成分自身对照法 当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。 (3) 内标法测定供试品中杂质的总量限度 采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。 如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。 (4) 内标法加校正因子测定供试品中某个杂质或主成分含量 按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。 再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。 当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。 (5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。[em61]
[color=#444444]实验室长久没用液相色谱了,型号是FL2000的,双泵,前两天在进样作分析的时候发现走色谱甲醇的时候 压力高达20多MPa,走了一上午才降到17Mpa,如果走流动相的话压力更高了,而且基线也很不稳定,像那种针刺一样的基线,请问各位大神们,该如何解决?[/color]
高效液相色谱法的计算方法高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。1.对仪器的一般要求所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。2.系统适用性试验按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子.(1) 色谱柱的理论板数(n)在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。(2) 分离度定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。(3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。(2) 主成分自身对照法当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。(3) 内标法测定供试品中杂质的总量限度采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。(4) 内标法加校正因子测定供试品中某个杂质或主成分含量按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。(5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。
液相色谱法中,内标法定量分析。有无要求内标物的峰面积和被测物的峰面积一样大小?有没有一定的范围要求?
各位大鱼大虾们好!我是新手,我们公司刚刚买了一台岛津的高效液相色谱LC-20A (配有荧光检测器和紫外检测器)用来测试双酚A(BP-A),目前我们用的是荧光检测器。我们现在设置的参数是这样的:激发波长为227nm,发射波长为313nm,流速为0.8ml/min ,流动相用的是纯乙腈,测试出来的峰有溶剂峰干扰。不知道是哪个环节设置出问题了,还请各位大鱼大虾多多指教。有点急,谢谢!
农药混剂分析方法,请指教,互相探讨[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=19740]福美双与腈菌唑高效液相色谱分析方法[/url][em04] [em04]
请介绍几款双泵以上的高效液相色谱仪的型号与报价!!/:| 急用!!

国产与进口液相色谱仪测试比对报告1.比对目的 选取2010年药典二部及美国药典中典型方法及食品添加剂国标方法对某国产品牌上海伍丰EX1600高效液相色谱仪及其他品牌(以waters仪器为主)液相色谱仪的各项性能进行比较,明确国产仪器优化与改进方向。2.比对依据与原理1. JJG705-2002 液相色谱仪计量检定规程2. EX1600高效液相色谱仪及其他品牌高效液相色谱仪使用说明书3. EX1600高效液相色谱仪系统适用性测试方案及综合改善测试方案4. GB/T 19681—2005食品中苏丹红染料的检测方法--高效液相色谱法5. VensuilAA氨基酸分析方法6. 2010年药典二部及美国药典中双氢青蒿素、可可碱与茶碱、维生素A及阿托伐他汀钙等相关物质的测定方法。7. 需比对的性能指标选取原理:与《JJG705-2002 液相色谱仪计量检定规程》要求直接相关;与仪器关键部件的技术指标直接相关;与仪器测定结果重复性,即仪器稳定性直接相关。8. 样品选取原理:测定结果直接反应仪器的稳定性;属于常用或者经典方法,单标、混标体系均有所涉及;等度和梯度方法均有所涉及(反映梯度误差指标);与检测器的基本波段(低、中、高波段)均相关的项目(反映全波段检测稳定性)。3.比对内容 高效液相色谱仪对样品的分离与测定结果好坏与仪器的稳定性及色谱柱的柱效相关,现使用同样的色谱柱进行EX1600高效液相色谱仪及其他品牌的色谱仪各项性能比对,就需要了解构成液相色谱的各个系统及整机的稳定性情况。就仪器各个系统的稳定性来讲,按照《液相色谱仪计量检定规程》测试方法,对流量准确度与重复性、噪声与漂移的测定结果能够从仪器方面直接反映输液系统及检测器的运行情况,明确指出这两个核心部件的优化与改进方向。而就整机稳定性来讲,还需要配合标准样品的测试,对其检出限、线性范围和梯度误差及定性定量重复性结果进行比较和评价,作为应用测试指标进一步反映仪器的稳定性好坏。将仪器性能指标与应用测试指标相结合,才能全面、客观地对仪器各项性能的稳定性进行评价与改进。现将主要测试内容列在下表:http://ng1.17img.cn/bbsfiles/images/2013/11/201311100229_476241_2369266_3.png 根据以上所列比对项目的比对内容,选择苏丹红ⅠⅡⅢⅣ、双氢青蒿素、阿托伐他汀钙、可可碱与茶碱、维生素A、氨基酸作为衡量输液系统、检测器、整机及软件性能综合指标优劣的标准测试样品,其特有的测定条件及测定结果可以综合反映色谱仪的稳定性,现将其优势测定条件与测定结果反映的综合指标列在下表并具体阐述如下:http://ng1.17img.cn/bbsfiles/images/2013/11/201311100231_476242_2369266_3.png 据食品国家标准、药典二部及美国药典中的典型方法,我们选取了近年来与食品、药品质量可控性、有效性及安全性密切相关的六类标准样品,依据其测试结果来衡量高效液相色谱仪各项性能稳定性。 苏丹红作为化工试剂,被不法商家非法添加入调味品中,是近年来食品安全领域的热点问题。苏丹红ⅠⅡⅢⅣ易致癌,是我国明文禁止的非食品添加剂。在相关食品国家标准中对其含量有具体的限量值,鉴于其实际测定中响应值低,我们对苏丹红的检测结果可直接体现高效液相色谱仪的噪声、漂移、检出限及线性范围(下限)等指标优劣。同时,苏丹红ⅠⅡⅢⅣ作为四元混标体系,国标方法规定苏丹红Ⅰ检测波长为478nm,苏丹红ⅡⅢⅣ检测波长为520nm,在检测过程中须转换波长,测定结果能够直接反映高波段、波长转换时的噪声与检出限;分离条件为梯度洗脱,可反映输液系统的梯度误差及双泵在梯度洗脱的稳定性。 双氢青蒿素作为单标体系,色谱测定条件为检测波长210nm,测定结果可直接反映检测器在低波段的稳定性及单泵运行情况。 阿伐他汀钙液相色谱检测波长为244nm,位于检测器优势波段的相对低波长处,流动相中含有高比例、洗脱能力较强的试剂四氢呋喃,可直接反映检测器优势波段噪声及单泵稳定性。 氨基酸混标体系共包含17种氨基酸组分,为复杂难分离体系,检测波长为254nm,测定时为梯度洗脱,测定结果可直接体现检测器优势波中段噪声及梯度洗脱时双泵运行稳定性。 可可碱与茶碱的二元混标体系,测定时检测波长为272nm,位于检测器优势波段的相对高波长处,可进行等度分离,测定结果直接反映了检测器优势波段稳定性及双泵在等度洗脱时的稳定性。 维生素A的高效液相色谱测定体系为正相色谱,流动相为正己烷:异丙醇[font=Tim
以下资料是转自其它网站:1960年代,由于气相色谱对高沸点有机物分析的局限性,为了分离蛋白质、核酸等不易气化的大分子物质,气相色谱的理论和方法被重新引入经典液相色谱。1960年代末科克兰(Kirkland)、哈伯、荷瓦斯(Horvath)、莆黑斯、里普斯克等人开发了世界上第一台高效液相色谱仪,开启了高效液相色谱的时代。高效液相色谱使用粒径更细的固定相填充色谱柱,提高色谱柱的塔板数,以高压驱动流动相,使得经典液相色谱需要数日乃至数月完成的分离工作得以在几个小时甚至几十分钟内完成。 1971年科克兰等人出版了《液相色谱的现代实践》一书,标志着高效液相色谱法(HPLC)正式建立。在此后的时间里,高效液相色谱成为最为常用的分离和检测手段,在医`学教育网搜集整理有机化学、生物化学、医学、药物开发与检测、化工、食品科学、环境监测、商检和法检等方面都有广泛的应用。高效液相色谱同时还极大的刺激了固定相材料、检测技术、数据处理技术以及色谱理论的发展。 1960年代前,使用的填充粒大于100μm,提高柱效面临着困境,后来的研究人员便采用微粒固定相来突破着一瓶颈。科克兰、荷瓦斯制备成功薄壳型固定相,这种在固定相在玻璃微球表面具有多孔薄壳,实现了高速传质,为高效液相色谱技术的发展奠定了稳固的基础。随着填料粒径的降低,更高的柱效也得以实现。 1960年代研制出气动放大泵、注射泵及低流量往复式柱塞泵,但后者的脉冲信号很大,难以满足高效液相色谱的要求。1970年代,往复式双柱塞恒流泵,解决了这一问题。1970年代后科克兰制备出全多孔球形硅胶医`学教育网搜集整理,平均粒径只有7μm,具有极好的柱效,并逐渐取代了无定形微粒硅胶。之后又制造出的键合固定相使柱的稳定性大为提高,多次使用成为可能。1970年后,适合分离生物大分子的填料又成为研究的热点。1980年后,改善分离的选择性成为色谱工作者的主要问题,人们越来越认识到改变流动相的组成事提高选择性的关键。







 我要推广仪器
我要推广仪器
 下载APP
下载APP