
液相色谱-串联质谱条件测定水果中的多农药残留方法优化本法建立了水果多种农药残留量的快速、简便、准确的测定方法,通过对样品前处理方法、仪器检测方法的考察优化,建立液相色谱-串联质谱检测方法。其中以日本制定的“肯定列表”中的“一律标准”最为严格,限量为 0.01mg/kg,而我国的残留限量标准还不够完善,很多农药还没有制定限量标准,其方法的测定低限(定量限)能达到 0.01mg/kg的要求。1、液相色谱条件考察:在方法建立过程中对液相色谱条件进行了考察,主要考察了色谱柱、流动相等。在色谱柱选择时,比较了BEH C18、 HSS T3、 Zorbax Eclipse Plus C18等色谱柱,发现HSS T3色谱柱对甲胺磷、乙酰甲胺磷等大极性农药的色谱保留效果较好,所以选择HSS T3色谱柱进行下一步的研究。在流动相考察时,发现在流动相中加入0.1%的甲酸可以改善多菌灵、噻菌灵等农药的分离,而且加入甲酸可以在电喷雾正离子( ESI+)模式电离时提供H+,提高电离效果,所以选择在流动相中加入0.1%的甲酸,比较了在水相和有机相中均加入0.1%的甲酸、仅在水相中加入0.1%的甲酸两种情况,发现色谱分离及质谱电离无显著差别,为简化操作和便于使用,选择仅在水相中加入0.1%的甲酸。流动相的有机相选择时考察了甲醇、甲醇-乙腈( 1+1, V/V)、乙腈三种情况,发现采用乙腈时色谱柱的柱压较低,色谱分离也较好,但毒死蜱、辛硫磷、特丁硫磷、敌敌畏等常用有机磷农药的质谱响应值低、重现性差,而选用甲醇时可以显著提高这些化合物的质谱响应及重现性,综合考虑后选择甲醇为流动相的有机相。由于此次分析的多农药的化学性质差别较大,从高极性到低极性均有分布,所以色谱分离时需要采用梯度洗脱模式,通过实验考察,最终确的液相色谱条件如下:a)色谱柱: HSS T3柱,长100 mm,内径2.1 mm,粒径1.8 μm,或相当者; b) 流动相:甲醇-0.1%甲酸溶液梯度洗脱,参见表 1。 https://ng1.17img.cn/bbsfiles/images/2020/09/202009211516176216_1219_2166779_3.png!w607x264.jpgc) 柱温: 35 ºC
在建立药物残留方法的时候,我们都是要用标准品溶液首先优化药物的质谱条件的,那这个时候流动相是如何选择的,Thermo的操作规程是要求使用50%甲醇50%水作为带动流动相,将蠕动泵注入的标准溶液带入质谱内,但我想也不至于是硬性规定吧,也不知道是否有依据。我做试验药物响应还可以,也是运气好啊,可是其他人做的药物按照这样做响应却很低,即使浓度配的很高,但是怎么调正离子模式响应也达不到e7,那遇到这样的问题应该如何去解决呢,按照这样优化,系统的压力不能高于多少。我想是不能太高的,不然药物很难被带入,是不是有硬性规定呢。其他厂家进行质谱优化的时候是不是也用的三通呢,AB的好像不是的,直接用蠕动泵打进去的,那还有其他的呢,比如Agilent,Waters,岛津的等等。有没有版友用过这些的,有什么体会,具体的操作是怎样的,能否详加列入。
我做有关物质的结构鉴定,优化好色谱条件后,质谱上却没有响应,由于刚刚开始做MS,想请教各位提高离子响应的方法有那些啊?
第一次用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],想用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]来定量,文章都会进行质谱条件优化,但是没说是怎么优化的?想在这里请教一下大神们!
![[原创]:TSQ质谱仪化合物条件优化标准操作规程](https://ng1.17img.cn/bbsfiles/images/2006/05/200605240732_18885_1237095_3.jpg)
论坛里技术性帖子较少,近期打算写一系列的帖子,关于TSQ[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用仪[/color][/url]的标准操作,大家喜欢的话,我会继续的。今天先谈谈TSQ质谱仪化合物条件优化标准操作规程。注:转载请注明来源及作者,谢谢!一、优化待测化合物ESI质谱条件1 样品导入方式的建立1.1 选择适当长度的Peek管将两端通过接头分别与液相系统和切换阀2号口相连。1.2 选择适当长度Teflon管将一端通过接头与切换阀1号口相连,并将另一端置于废液瓶中。1.3 选择适当长度的Peek管将一端通过接头与切换阀3号口相连,另一端通过三通分别与离子源和样品转移毛细管相连。1.4 将200 uL左右样品溶液吸入250 uL进样注射器中。1.5 将进样注射器通过一个接头和一个二通与样品转移毛细管另一端相连。1.6 按住注射泵黑色释放钮将注射泵手柄升高。1.7 将进样注射器小心置于支架上并将注射泵手柄下移至进样注射器活塞柄顶端。2. 质谱条件优化步骤2.1 在Tune Master界面点击On/Standby激活质谱仪。2.2 选择离子极性模式(正离子或负离子),如需进行正负离子切换,将将Spray Voltage调至0后操作。2.3 进入Compound Optimization Workspace。2.4 在Define Scan窗口选择Q1MS扫描模式和Full Scan扫描类型。2.5 在Optimize Compound Dependent Devices窗口设置下列参数: Spray Voltage设为3500 V Sheath Gas Pressure设为30 arb Aux Gas Pressure设为10 arbCapillary Temperature设为350℃Source CID设为0 V2.6 激活注射泵以5 uL/min流速将进样注射器中的样品溶液导入质谱仪。2.7 激活液相色谱泵选择适当流速将流动相导入质谱仪,观察到待测化合物的准分子离子峰峰强度在10的6次方左右,否则增大进样流速或选用浓度更高的待测化合物溶液(样品浓度一般建议1-10ug/mL,建议用甲醇或乙腈溶解)。2.8 在Compound Optimization界面显示Single Sample窗口,选择MS Only优化模式和Syringe Pump Infusion入口类型选项。2.9 优化Tube Lens Offset、Spray Voltage、Sheath Gas Pressure、Aux Gas Pressure和Source CID获得待测化合物稳定的准分子离子峰。2.10记录并保存准分子离子质谱图。2.11选择MS+MS/MS优化模式设置Parent Mass、Charge State和Num Product对子离子进行优化,优化前完成下列设置: 将Source CID设为0 V 将Collision Pressure设为1.5 mTorr将Quad MS/MS Bias设为-1.0 V2.12接受Collision Energy优化结果,并将Source CID设为优化值(由2.9得到)。2.13记录并保存子离子全扫描质谱图。2.14保存Tune Method文件。3. 注意事项3.1 待测化合物溶液浓度为1-10ug/mL3.2 改变流动相比例和流速后应重新进行对Sheath Gas Pressure和Aux Gas Pressure进行优化3.3 手动优化Capillary Temperature3.4 点击Start开始自动优化程序,优化结束时点击Accept接受优化结果,或者点击Undo后再点击Accept保持优化前的仪器配置3.5 仪器常用参数设置见下表。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605240732_18885_1237095_3.jpg[/img]
那位大侠液质配的是国产蠕动泵?我也想购买一个,做质谱调谐优化条件用。请把您用的品牌和型号以及用的效果如何告诉我好吗?先谢了。
就是洗脱溶剂、洗脱流速等等,是不是先直接连质谱,优化好了这些条件才一起接六通阀和分析柱做柱切换?
仪器:Varian [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/ms 4000,在进行方法开发时,不知如何利用软件AMD对ms/ms质谱条件进行优化 ,主要是想降低方法检测限
请问大家在用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]建立分离分析方法时,都优化哪些条件,从哪些方面考虑的呢?多谢!
各位大佬你们好啊,请问[font=-apple-system, BlinkMacSystemFont, &][color=#121212]Orbitrap Fusion? Lumos? 型号的质谱,有什么质谱参数优化的方法,可以用来提高谱图鉴定量,跪求!!![/color][/font]
http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif当你拿到一个标准后,里面的液相条件和质谱条件你还会进行摸索吗?还是只按照上面的条件设置?还有就是,前处理按照标准的方法做多少次你会判定此方法回收率做得好与坏?曾经做过一个兴奋剂的标准,质谱条件是别人摸索好的方法,前处理是自己优化出来的,大概做了十几次回收率还是50---60%,最后判断这方法如果不加内标绝对做不到像标准上说的那样1ng/ml回收率在70%---120%
如题,请问我这边买的标准品已经是代谢物了,还需要经过衍生化后才能上质谱优化吗?还是可以不衍生直接用质谱优化?
各位老师好,肉桂酸标品我[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测不出来,400ng/ml标准品时响应极低。 导师认定我条件没优化好,工程师不建议优化质谱源参数,为了仪器干净我只用甲醇乙腈和不同比例的甲酸优化流动相(文献也有用液相或[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做出来里的),我除了液相色谱的优化还能做什么嘛??现在感觉还是要优化质谱参数,但不知道优化什么(???.???)????图一先质谱参数,图二0.1甲酸乙腈-0.1甲酸水,柱温30 梯度洗脱[img]https://ng1.17img.cn/bbsfiles/images/2022/02/202202121438165640_2049_4210178_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/02/202202121438167954_971_4210178_3.png[/img]
老师们好,最近在进行指纹图谱的相关实验,产生了一个疑问,希望老师们能帮忙解答一下。在实验中,应该先优化样品前处理的条件还是色谱分析的条件呢?若先优化前处理条件,该用什么色谱分析方法对处理好的样品进行分析呢?若采用的色谱分析方法和最后优化的色谱方法不同,会不会导致优化时变量不唯一的情况?
目前在做质谱参数优化,4个化合物,均为卤代甲苯类化合物。仪器为Agilent 6470QQQ,scan模式下正负离子扫描,化合物紫外吸收很好,但其TIC图均未出现明显峰形,提取EIC图也没有峰。同事说该类化合物很难离子化,后试过将水相改为0.1%FA溶液及10mmol/L乙酸铵溶液,促进离子化。只有一个物质在10mmol/L乙酸铵溶液条件下得到加钠峰的EIC图,但与其对应的紫外吸收峰时间间隔了1min,样品从紫外直接流入质谱检测,一般情况下0.2-0.3min就可以完成,因此对此存在质疑。大家碰到过这种情况吗?或是有什么建议吗?
五氯酚在质谱里的条件 峰只有264.7那个离子的峰好看一些 其他三个离子的峰都惨不惹睹
老师们好,最近在做分析的过程中发现了一个问题,困扰我许久,希望各位老师能帮忙解答一下。如题,在建立指纹图谱的时候,不知道是该先优化样品前处理的方法,还是先优化色谱分析的方法?若先优化前处理方法,处理好的样品该在什么色谱条件下进样分析呢??若是处理好样品先找一个色谱分析条件进样分析,后续优化色谱条件后发现最优的分析条件不是优化前处理方法时所用的进样条件,岂不是会造成优化条件时的变量不唯一吗?还望各位老师解答,感谢??
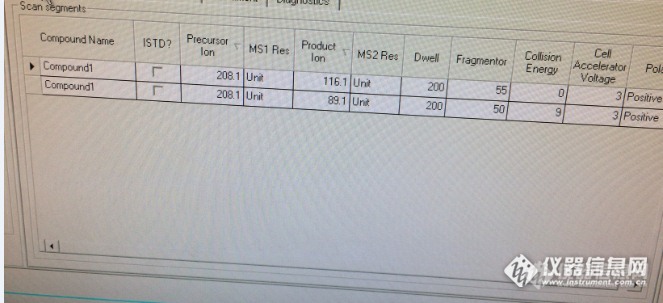
安捷伦1290-QQQ6430上优化涕灭威母离子 208 :1.做Scan时找不到母离子,这时候我们需要把毛细管电压正模式和负模式都降到1000去,这样做全扫找到了母离子。http://ng1.17img.cn/bbsfiles/images/2016/03/201603051303_586080_2779413_3.png这两个值改为10002.锁定母离子后就去找子离子这时候需要增加响应,所以把毛细管电压调回4000 35003.找子离子,裂解电压默认值一般是135,但因为涕灭威比较容易碎裂,所以我们裂解电压设置为 20 30 50 60 就可以了,这模式中可以添加四个扫描范围。4.顺利找到涕灭威的两个子离子 116.1 89.1 ,满足四分要求。5.接着优化它的裂解电压 碰撞能量 加速电压。结果 http://ng1.17img.cn/bbsfiles/images/2016/03/201603051310_586082_2779413_3.png裂解电压和能量都和其它物质差别大,116.1的碰撞能量竟然是0的时候响应最高。1290-QQQ6490的优化情况这个优化流程和上面一样,但是6490的参数比较多 通过优化具体参数如下:干燥器温度150 流速18L/min 鞘气温度350 鞘气流速12L/min 毛细管电压+3500 , -2000喷嘴电压+ 500. -1000 分子漏斗 +高端70 低端40 ,- 高端90低端60 这里的分子漏斗电压只适合做涕灭威一个时得到响应更高 检测浓度0.1ng/mL完全无压力。如果需要同时和其它物质一起检测 分子漏斗只要改回 +高端150 低端60 ,即可。但是EMV+电压需要加200最后优化结果 母离子 208.2 子离子89.1 115.8 碰撞能量分别是10和0,裂解电压都为380(仪器不可调),加速电压为5总结下:6430毛细管电压可以影响响应,但是太高时可能找不到母离子,原因分析物质容易碎裂,需要降低电压才能找到母离子。6490设计和6430不一样,有分子漏斗需要优化,对于易碎裂的物质需要把电压降低。而且碰撞能量是0,优化时,需要注意。如你们喜欢,可以加入我们的小团队。 名为:shooter 团队里面有大学的教授(帅气的老外教授(我们的最终决策人)),各个地方的检验人员,和对液相感兴趣的年轻朋友。我们团队现在的进程是讨论液相色谱的条件,通过我们来把一些在液相上分析时间长的旧方法改为快速高效的方法(必须要成为实例)。 希望你们的加入,具体方法在论坛留言给我,我会尽快回复你们。 我们需要的你是能和我们融合为一个Team!
时间紧任务重,所以想优化下642的分析时间。原来做一个样,质谱分析40分钟,现在把程序升温改了,缩短到了了20分钟。内标也都提前出来了。但是峰面积逐个递减。不太懂怎么回事了。求助一下大神。原来是40℃保持2分钟,5℃每分升到120,保持两分钟,再10℃每分升到230,保持九分钟,大概是这样。现在是40℃保持2分钟,10℃每分升到200保持两分钟。[img]https://ng1.17img.cn/bbsfiles/images/2023/03/202303101804527052_8694_4020217_3.png[/img]

安普SPE方法开发及条件优化课程今天在广州开课,有没有版友参加呀[img=,595,714]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090902_01_2946960_3.jpg!w595x714.jpg[/img]

药典方法红花含量测定使用两种方法分别测定羟基红花黄色素A和山奈素两种成分含量色谱条件如下:[img=,477,96]https://ng1.17img.cn/bbsfiles/images/2020/12/202012101605485344_5670_2204446_3.png!w477x96.jpg[/img]如果用一种流动相同时测定两个组分,可分别采集403nm,367nm两个通道信号。色谱条件如何优化?
有没有做瘦肉精的,请问下waters[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用仪[/color][/url]上优化的喷布特罗的质谱方法是啥?差这一个质谱方法,感谢感谢!
因工作需要且初次接触热分析仪器,急需有关“热分析方法及其条件的选择与优化”方面的资料,谢谢啦!

[align=left][font='times new roman'][size=16px]三重串联四极杆[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url] /质谱联用测定蔬菜和茶叶中 49 种农药残留[/size][/font][size=16px]的方法优化[/size][/align][align=left][font='times new roman'][size=16px]以前样品经提取、净化后,用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]进行定性定量分析。或者用GC-FPD检测器测定有机磷,GC-NPD检测器测定氨基甲酸酯和有机磷,用GC-ECD检测器测定有机氯和拟除虫菊酯。一般需分组,如无[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url],对检出峰重叠的农药要求双柱定性。GC和[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]检出限一般为几个到几十个μg/kg。[/size][/font][/align][align=left][font='times new roman'][size=16px]三重串联四极杆QQQ :第一质量分析器(Q1、四极杆1)选择某一质核比的离子进入碰撞池(Q2、六极杆2),被选择的离子在池里与碰撞气体(氮气)碰撞,经过碰撞诱导解离(CID)产生的子离子(碎片)由第二个质量分析器(Q3、四极杆3)进行扫描分析。[/size][/font][/align][align=left][font='times new roman'][size=16px]单四极杆质谱中,离子监测模式(SIM)只监测保留时间范围内的少数几个离子,和全扫描模式有相同的杂质干扰,GC-QQQ可以大幅度降低甚至消解影响SIM方法准确度和检测限的基质干扰, MRM的检测是基于次级“碎片离子”,由第一个四极杆Q1产生的分析物的母离子在Q2经六级杆碰撞解离而产生,与SIM比有同样的选择性,但能保证至少有一个是由母离子特有而非干扰物产生的,基线漂移明显减少。在Q1的质谱过滤过程中,样品中所有低质荷比的离子都被过滤掉,产生的唯一“碎片离子”在“零噪声”中进行检测,得到“干净”的色谱图,对复杂的基体样品也能很好的定量。使得MRM检测限更低。图[/size][/font][size=16px]1[/size][font='times new roman'][size=16px]~[/size][/font][size=16px]2[/size][font='times new roman'][size=16px]为蔬菜和茶叶色谱图。茶叶提取物是最复杂基体样品之一,然而GC/MS/MS的提取离子流图却很“干净”,对复杂的基体样品也能很好的定量。[/size][/font][/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310310826220157_7469_4033901_3.png[/img][/align][font='times new roman'][size=12px]图[/size][/font][size=12px]1[/size][font='times new roman'][size=12px] 含有10ppb β-六六六的蔬菜加标样品GC/MS/MS提取离子流图和MRM transition离子图[/size][/font][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310310826222061_4447_4033901_3.png[/img][font='times new roman'][size=12px]图[/size][/font][size=12px]2[/size][font='times new roman'][size=12px] 含有10ppb杀螟硫磷绿茶样品GC/MS/MS提取离子流图和MRM transition离子图[/size][/font][align=left][font='times new roman'][size=16px]质谱条件的优化 [/size][/font][font='times new roman'][size=16px]:[/size][/font][font='times new roman'][size=16px]为获得最佳的质谱条件保证农药定性、定量的准确性,对分析物的母离子、子离子以及碰撞能量等一系列质谱参数进行优化。[/size][/font][/align][align=left][font='times new roman'][size=16px]首先进行全扫描,得到母离子和保留时间。取适宜浓度的标液在扫描范围50D~600D进行。母离子选择应选高的质量数(定性)、高的丰度(灵敏度)。第二步做产物离子扫描(production scan)选择子离子和碰撞能量(collision energy)。分时间段,5~40 V范围内,以5V为跨度,对已选母离子进行碰撞能量的优化,子离子必须有足够的丰度,可以用于痕量分析,灵敏度好,选择性要高,不要选择会被干扰的子离子,根据欧盟法规,至少需要一个母离子和两个子离子以满足4点鉴定法。见图[/size][/font][size=16px]3[/size][font='times new roman'][size=16px]~[/size][/font][size=16px]5[/size][font='times new roman'][size=16px]。第三步多反应监测MRM的运用。[/size][/font][/align][img]" style="max-width: 100% max-height: 100% [/img][font='times new roman'][size=12px]图[/size][/font][size=12px]3[/size][font='times new roman'][size=12px] [/size][/font][font='times new roman'][size=12px]母离子在不同的碰撞能量(5~20V)下的碎片[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]图[/size][/font][img]" style="max-width: 100% max-height: 100% [/img][font='times new roman'][size=12px]图[/size][/font][size=12px]4[/size][font='times new roman'][size=12px] 母离子在不同的碰撞能量(5~40V)下的碎片离子叠加质谱图[/size][/font][align=center][img]" style="max-width: 100% max-height: 100% [/img][/align][align=center][font='times new roman'][size=12px]图[/size][/font][size=12px]5[/size][font='times new roman'][size=12px] 农药混合标准MRM28min~29.2min色谱放大图(亚胺硫磷)[/size][/font][/align][align=left][font='times new roman'][size=16px]通过对柱温(程序升温)、载气流速、进样口以及质谱参数等条件的优化,[/size][/font][font='times new roman'][size=16px]蔬菜和茶叶中[/size][/font][font='times new roman'][size=16px]氨基甲酸酯、有机磷以及有机氯和拟除虫菊酯4大类49种农药分离效果、峰形、耗用时间的综合情况最好,总时间为37.867 min,能获得理想的最低检出限,可以满足实际测试的要求。总离子流图(TIC)见图[/size][/font][font='times new roman'][size=16px]6[/size][/font][font='times new roman'][size=16px]。[/size][/font][/align][align=left][img]" style="max-width: 100% max-height: 100% [/img][/align][align=left][font='times new roman'][size=12px]图[/size][/font][font='times new roman'][size=12px]6 [/size][/font][font='times new roman'][size=12px] [/size][/font][font='times new roman'][size=12px]农药混合标准(10μg/kg)的GC/MS/MS总离子流图[/size][/font][/align]
有谁做过食品中红曲红素的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]方法,麻烦告诉我下质谱条件,谢谢。
我现在做一个生物碱的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS的定量,质谱条件优化的挺好,可是色谱条件不怎么理想,出的TIC图峰宽太宽,有点拖尾。用的是10厘米的短柱,流动相是45%ACN和55%水(0.1%甲酸和10mM醋酸铵),流速0.3希望各位大虾帮帮忙,给点建议,这个HPLC条件怎么优化的好点,本人刚入行,很多都不是很清楚
本人做激素的检测,目前是在运行单标逐一优化液相条件(质谱条件已经确定并且优化完毕),色谱柱:安捷伦C18柱流动相A:水(含有0.1%的甲酸),B:甲醇。梯度:0-6min(35%B-40%B),6-12min(40%B-80%B),12-15min(80%B-95%B),15-15.1min(95%B-35%B),15.1-19.1min(35%B保持4min)。现在刚做完16个样品,只有一个的峰型不好,稍微有一点儿拖尾,但是又感觉像是没有分开(图1),问题1:这个没有分开的怎么处理能好一些?(但是在质谱定量分析软件中,定量子离子不受影响,可是定性子离子就不好了,怎么办?有影响吗? 图2)问题2:16个样品的出峰时间都集中在12-15.1min之间,有几个的保留时间都是14.3,怎样才能让他们再分开一些,也就是为了美观好看,另外我想让他们时间都往前提一下,十几分钟感觉太晚了, 希望提供一个参考的梯度。 谢谢PS:我感觉柱子的问题不大,我是这么想的16个样品15个都没出现问题,单单这一个,柱子的影响因素不大。
条件优化后保留时间变化,因为杂峰一直较多,现在找不到目标峰了。是按文献方法优化的,但走出来的峰都没有和文献离子谱图一致的。与优化前的离子谱图也不一致。不知道是什么原因,峰没出来?
做单组分主要是用作预实验,但是在看文献的过程中有点搞不懂条件优化的顺序,条件包括,SPE柱的选择、SPE柱的流动相的种类及比例、分析柱的选择、分析柱的流动相的种类及比例、质谱条件……我的疑惑是,柱子还没确定,怎么去做后面的呢?我看到文献里,在【优化SPE柱条件】的时候,得用一个确定的流动相条件【后面分析柱的流动相条件】去看峰形是否拖尾来选择保留和吸附能力都比较好的作为后面分析的柱子,那分析柱的流动相条件又是什么时候优化的呢?其它的条件也是一样,还没确定这个条件的时候,怎么知道要用哪个种类和比例的流动相去做,确定另一个条件呢?所以不太清楚,实际到底应该怎么做?谢谢!
实验室有一台安捷伦G6120的液质联用仪,没有配置液相检测器,配一台ESI/APCI的质谱。1请问如果我想用外标法该怎么定性定量?2还有必要优化色谱条件吗?3质谱条件该怎么优化?







 我要推广仪器
我要推广仪器
 下载APP
下载APP