色谱出的数据因为要求要按百分含量,在数据进行归一的时候,色谱所出数据有时候只有七八十,但归一后总数有一百对吗?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析中鬼峰产生的原因及排除方法?
已经不怎么用液相色谱了,整理资料发现自己还有留着一些厂家的数据库,虽然过时了,但没总结,所以现在写点东西,记录一下。这些从哪来的呢?都是厂商的广告和宣传品,用的都是自己的产品,但是对做实验还是有参考价值的。数据库包括:安捷伦色谱应用方法数据库、默克的色谱应用大全和瓦里安的SCANVIEW8.0我的安捷伦的色谱应用方法数据库里是2005年的分为气相和液相两类,方法不是很多,只由65M,6百多张图,基本在它的宣传资料上都有,但做成数据库,可以方便的检索,重要的是它用中文作目录,可以中文检索,方便英文不好的同仁看看。默克的色谱应用大全叫Chromcircle 2004,不知道是不是2004年出的,通过这个我才知道除试剂外,默克跟液相色谱柱也有关系(做填料)总共286M,宣传中说里面包含了3500篇HPLC应用报告,还有7700篇TLC的应用文摘,但它做成了数据库,不知道到底真的有多少种。全英文检索。以上两种都可以直接使用瓦里安的SCANVIEW8.0应该是2005年出的,需要安装后生成一个十几兆的程序,可以检索,但看图时提示我要插入光盘,后来我发现不需要虚拟光驱,只要把盘里面的文件复制到硬盘上,再安装,当要看图时,它会从原来的文件夹中调出来,程序共计304M,里面的pdf方法是247M,4238个文件,也是全英文的后两种本来我准备压成40多M的几个文件上传,经过了两天,一直失败,过几天看我有没耐心把它割成20多M的压缩文件上传
1.关于色谱积分,想请教各位大神,什么样的情况可以采用谷到谷积分,又在什么样的情况下可以采用垂线积分,有大神知道具体依据没?如果出现一个小的杂质峰,但是附近基线走的不好(有点有点像双峰的感觉),又该如何合理积分?2.关于数据处理,按照GMP要求:是不是每次数据处理方法必须要一致,不允许有任何改动?如果是,具体原则是什么,出于药典哪条法规?3.峰识别,系统溶液中各组分峰的保留时间与样品中相应组分峰之间的保留时间相差多少,是可以被接受的?这个问题同样站在审计角度!
请问一下大家,我用顶空-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]归一化法定量分析乙酰丙酸脱羧后丁醇和丙醇的产率,得到最后产率数据,那怎么根据数据作图??

【专家讲座】:如何确保实验室色谱数据的合规性【讲座时间】:2016年03月10日 10:00【主讲人】:沈晓峰 赛默飞应用专家,先后就职于制药行业与分析仪器行业,在药物分析、色谱分析、色谱数据系统技术支持等各相关领域已有二十年的经验。现主要负责支持色谱数据系统并针对客户需求提供相应的合规解决方案。【会议简介】报告结合当前形势,解读国内外的最新法规要求,通过实例详细介绍使实验室色谱数据合规的解决方案。帮助您在遵循法规要求的前提下快速、有效地得到结果。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名,通过审核后即可参会。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2016年03月10日 9:304、报名参会:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/17815、报名及参会咨询:QQ群—171692483http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667142_2507958_3.jpg
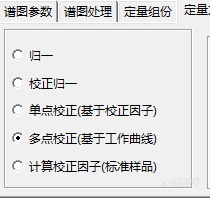
[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分析方法提供了以下五种方法:[img=,210,198]https://ng1.17img.cn/bbsfiles/images/2022/04/202204241156010412_9653_3138643_3.png!w210x198.jpg[/img]其中归一法和校正归一法有什么区别?是不是归一法不用校正因子,而校正归一法要用校正因子?但是要求校正因子就得做用标准物质算,上面求出的校正因子是绝对校正因子还是相对校正因子?算出校正因子,用归一法和多点校正(基于工作曲线)两种方法计算结果,相差比较大,不知道是什么原因
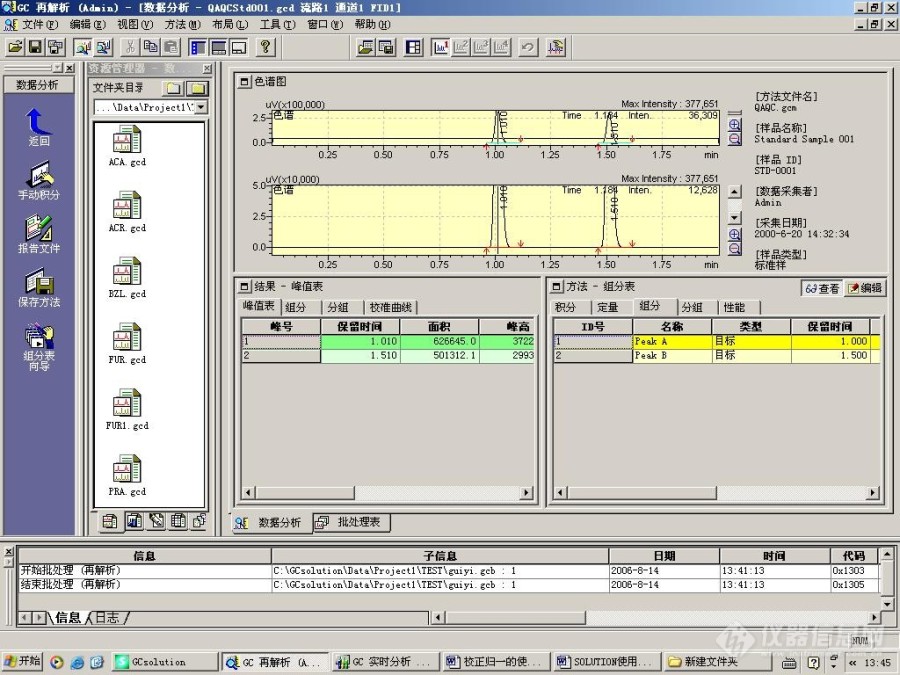
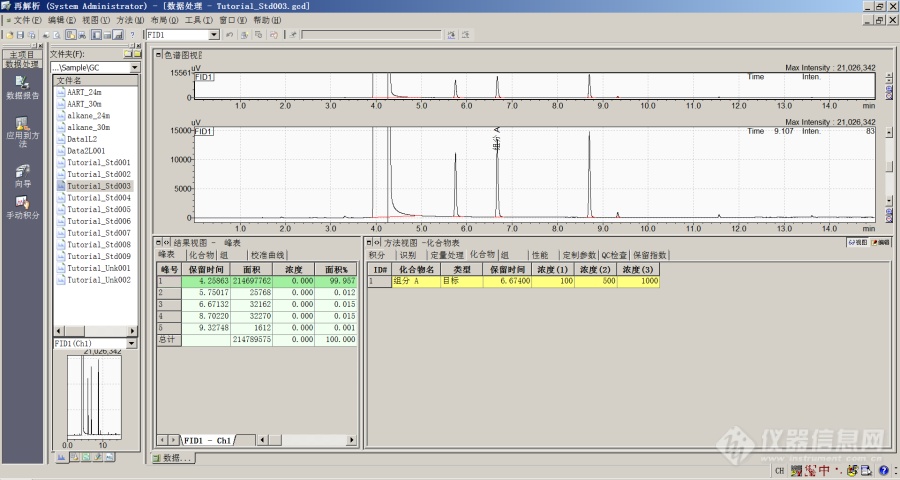
[align=center][font=宋体]用[/font][font=宋体][font=Times New Roman]GC[/font][/font][font=宋体][font=Times New Roman]SOLUTION [/font][/font][font=宋体]色谱数据工作函[/font][font=宋体]软件做校正归一[/font][font=宋体]定量[/font][font=宋体]的方法[/font][/align][align=center][font=宋体] [/font][/align][align=center][font=宋体][font=宋体]方法[/font][font=Times New Roman]1 [/font][font=宋体]—— 利用标准品,工作站自动测定校正因子[/font][/font][/align][align=center][font='Times New Roman'] [/font][/align][font=宋体][font=宋体]([/font] [/font][font=宋体]例如[/font][font=宋体]:使用标准样文件[/font][font='Times New Roman']QAQCStd001.gcd[/font][font=宋体]、[/font][font='Times New Roman']QAQCStd00[/font][font=宋体][font=Times New Roman]4[/font][/font][font='Times New Roman'].gcd[/font][font=宋体][font=Times New Roman],[/font][/font][font=宋体]和[/font][font=宋体] [/font][font='Times New Roman']QAQCStd00[/font][font=宋体][font=Times New Roman]7[/font][/font][font='Times New Roman'].gcd[/font][font=宋体] [font=宋体])[/font][/font][font=宋体][font=Times New Roman]1 [/font][font=宋体]打开数据文件[/font][/font][font=宋体][font=宋体]打开色谱数据再解析,点击屏幕左侧辅助栏中的[/font][font=宋体]“数据分析”图标,然后点击“打开”按钮,打开任意一个标准样品数据文件,观察[/font][/font][font=宋体]并记录待测[/font][font=宋体]物质的保留时间。[/font][align=center][font='Times New Roman'][/font][img=,529,397]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242308554443_8618_1604036_3.jpg!w690x517.jpg[/img][/align][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]1 [/font][/font][/align][font=宋体][font=Times New Roman]2 [/font][font=宋体]编辑和保存方法文件[/font][/font][font=宋体][font=宋体]点击屏幕右下角[/font][font=宋体]“方法”中的“编辑”;[/font][/font][font='Times New Roman'][/font][font=宋体]选择好“积分”项中的参数。[/font][font='Times New Roman'][/font][font=宋体]再点击“定量”。定量方法选择“校正面积归一”;计算依据[/font][font='Times New Roman']—[/font][font=宋体][font=宋体]“面积”;标准曲线级别[/font][font=Times New Roman]---[/font][font=宋体]“[/font][font=Times New Roman]1[/font][font=宋体]”;校准曲线类型—“线性”;零点[/font][font=Times New Roman]----[/font][font=宋体]“强制通过”;时间窗[/font][font=Times New Roman]----[/font][font=宋体]“[/font][font=Times New Roman]10-20%[/font][font=宋体]”。[/font][/font][font='Times New Roman'][/font][font=宋体][font=宋体]再点击[/font][font=宋体]“组分”,再组分表中填入“样品名称”、“保留时间”、“浓度”。[/font][/font][font='Times New Roman'][/font][font=宋体]再点击“查看”。[/font][font='Times New Roman'][/font][font=宋体]点击“文件”[/font][font='Times New Roman']—[/font][font=宋体]“方法另存为”,保存方法文件。[/font][img=,529,397]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309049176_5008_1604036_3.jpg!w690x517.jpg[/img][font=宋体] [/font][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]2[/font][/font][/align][font=宋体] [/font][img=,553,415]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309114845_8768_1604036_3.jpg!w690x517.jpg[/img][font=宋体] [/font][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]3[/font][/font][/align][font=宋体] [/font][img=,553,415]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309174022_9207_1604036_3.jpg!w690x517.jpg[/img][font=宋体] [/font][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]4[/font][/font][/align][font=宋体] [/font][img=,553,415]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309239367_2841_1604036_3.jpg!w690x517.jpg[/img][font=宋体] [/font][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]5[/font][/font][/align][font=宋体] [/font][font=宋体] [/font][font=宋体] [/font][font=宋体] [/font][font=宋体] [/font][font=宋体][font=Times New Roman]3 [/font][font=宋体]利用批处理计算结果[/font][/font][font=宋体][font=宋体]点击[/font][font=宋体]“批处理”,在批处理表中填入“样品名称”、“样品类型(标准样)”、“方法文件”、“数据文件”[/font][font=Times New Roman].[/font][/font][font=宋体] [/font][img=,553,415]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309299798_4494_1604036_3.jpg!w690x517.jpg[/img][font=宋体] [/font][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]6[/font][/font][font=宋体] [/font][font='Times New Roman'][/font][/align][font=宋体]再保存批处理文件。[/font][font='Times New Roman'][/font][align=center][img=,553,415]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309349240_9360_1604036_3.jpg!w690x517.jpg[/img][font='Times New Roman'][/font][font=宋体][font=宋体]图[/font][font=Times New Roman]7[/font][/font][font='Times New Roman'][/font][/align][font=宋体][font=宋体]然后点击绿色按钮[/font][font=宋体]“开始”。[/font][/font][font='Times New Roman'] [/font][font=宋体]运行批处理文件。[/font][font='Times New Roman'][/font][font=楷体_GB2312]此时可以将标准样品文件和未知样品文件加入批处理表中。[/font][font=宋体][/font][font=宋体][font=Times New Roman]4 [/font][font=宋体]查看定量结果[/font][/font][font=宋体][font=宋体]返回[/font][font=宋体]“数据分析”状态,打开批处理表中已经处理过的样品数据文件。即可观察结果。[/font][/font][font='Times New Roman'][/font][img=,553,415]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309406638_5854_1604036_3.jpg!w690x517.jpg[/img][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]8[/font][/font][/align][align=center][font=宋体] [/font][/align][align=center][font=宋体] [/font][/align][align=center][font=宋体][font=宋体]方法二[/font] [font=宋体]利用预先获得的相对校正因子[/font][/font][/align][font=宋体]方法二较为简单,在方法的组分表中直接输入组分的名称、保留时间和相对校正因子即可。[/font][align=center][img=,529,397]https://ng1.17img.cn/bbsfiles/images/2023/10/202310242309471866_5718_1604036_3.jpg!w690x517.jpg[/img][font='Times New Roman'] [/font][/align][align=center][font=宋体][font=宋体]图[/font][font=Times New Roman]9[/font][/font][/align][align=center][font=宋体] [/font][/align][align=center][font='Times New Roman'][/font][/align]

【专家讲座】:如何确保实验室色谱数据的合规性【讲座时间】:2016年03月10日 10:00【主讲人】:沈晓峰 赛默飞应用专家,先后就职于制药行业与分析仪器行业,在药物分析、色谱分析、色谱数据系统技术支持等各相关领域已有二十年的经验。现主要负责支持色谱数据系统并针对客户需求提供相应的合规解决方案。【会议简介】报告结合当前形势,解读国内外的最新法规要求,通过实例详细介绍使实验室色谱数据合规的解决方案。帮助您在遵循法规要求的前提下快速、有效地得到结果。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名,通过审核后即可参会。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2016年03月10日 9:304、报名参会:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/17815、报名及参会咨询:QQ群—171692483http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667144_2507958_3.jpg
[color=#444444]咱们实验室才买了一个伯乐的糖柱 87p, 2W多的那款测纤维素糖的,配着保护柱用了3个月,现在出现了鬼峰。鬼峰出现的地方在13-16.5分钟之间,刚刚好我的木糖和阿拉伯糖的样品出峰时间也在这,所以很糟糕。[/color][color=#444444]按照说明说上30%乙腈水0.1ml/min倒冲了一周了,鬼峰面积减小了,可鬼峰还是有,求大神提供彻底的解决方法。说实话,我实在不清楚为什么我测糖标准品也能把柱子给污染了。也不知道是不是保护柱的问题。[/color][color=#444444]附上空白试剂纯水的色谱图。[color=#444444]附上说明:肯定不是自动进样器污染,因为岛津的工程师已经帮我检查过了;肯定不是流动相污染,哇哈哈脱气后,每天更换;肯定不是样品污染,样品每天都是新配的。[/color][/color][color=#444444]或者如果送修的话,请大家谁能告诉我送修的单位?千万别说再买一根,2W多的柱子,我们实验室是要让我赔的。[/color][url=http://muchongimg.xmcimg.com/data/bcs/2017/0612/bw178h4350516_1497259738_276.jpg#opennewwindow][img]http://muchongimg.xmcimg.com/data/bcs/2017/0612/bw178h4350516_1497259738_276.jpg[/img][/url]
[color=black]Labsolution色谱数据工作站的系统适应性评价功能的使用方法[/color][align=center][color=black]概述[/color][/align][color=black]Labsolution色谱数据工作站系统适应性功能的使用方法[/color][align=center][color=black]背景介绍[/color][/align][color=black]色谱工作者在进行样品检测之前,通常需要确定色谱系统是有效和适用的,通常会使用分离度、柱效、拖尾因子和重复性来进行确认,对于医药行业的色谱工作者而言,这项要求尤其重要。[/color][color=black]目前实验室常见的色谱数据工作站均可以在色谱数据结果中出具分离度、理论塔板数、拖尾因子和重复性的数值,色谱工作者可以根据色谱数据工作站给出结果判定色谱系统适用性是否满足分析方法要求。[/color][color=black]使用Labsolution 系列色谱数据工作站的用户,可以利用工作站进行结果的评价。[/color][align=center][color=black]操作方法[/color][/align][color=black]例如某次实验分析获得三个标准数据文件——Std-001、Std-002、Std-003,分析方法对色谱数据中某组分的分离度要求大于1.5、拖尾因子要求小于1.6、理论塔板数要求大于2000。那么可以按照如下的顺序对数据文件进行处理:[/color][color=black]1 编制化合物表[/color][color=black]运行Labsolution的再解析模块,然后点击“数据处理”,打开任意一个标准数据文件,然后点击助手栏的“向导”图标,输入基本定量信息并编辑化合物表,如图1所示:[/color][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109212302334935_8950_1604036_3.png[/img][align=center]图1 化合物表视图[/align]2 编辑系统适应性计算信息[color=black]点击Labsolution色谱数据工作站菜单栏“方法”——“系统适应性设置”按钮,以理论塔板数为例,参照图2进行设置并保存成为方法文件(注意保存时一定需要选中“系统适应性设置”)。[/color][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109212302340002_7710_1604036_3.png[/img][align=center]图2 系统适应性设置[/align][color=black]3 编辑批处理表[/color][color=black]点击Labsolution色谱数据工作站的“批处理再解析”图标,编辑批处理表格,将需要进行计算的方法文件和数据文件添加到表格中,此外需要在批处理表格中添加系统适应性项目,并指定开始和结束行。[/color][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109212302342639_4066_1604036_3.png[/img][color=black]然后运行批处理表,即可得到系统适应性的评价报告。[/color][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109212302344036_2347_1604036_3.png[/img]
其他讲座资料看[url=http://www.instrument.com.cn/bbs/detail.asp/threadid/1679222/forumid/25/year/2009/query/search] 学习[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]跟yuen72老师入门[/url]定性,通常是指确认未知物质的组成。对于色谱,则一般指确认色谱峰的归属,因为色谱本身不具备定性能力。而对于色谱的数据处理,这里的定性则是在已经知道峰归属的条件下,如何在多次分析的保留时间有微弱变化的情况下,仍然确保正确的对色谱峰进行归属。例如,正常标定色谱,乙烷色谱峰出在5.2分钟。再次标定时,乙烷峰出在了5.1分钟。事实上,在标定和分析中,由于各种原因,组分的保留时间发生微弱的变化是很常见的。那么对于很机械的数据处理机和工作站,是如何确认保留时间变化了的色谱峰,是新组分,还是原组份呢?色谱工作站是用允许偏差来控制峰定性的。例如,设定乙烷的出峰时间为5.2分钟,对5.2分钟左右一定允许偏差内的色谱峰,都认为是乙烷。例如这个偏差设置为0.2分钟,那么在5.0至5.4分钟内的色谱峰,都认为是乙烷。我们都知道,偏差的表示方法有两种,一种绝对偏差,另一种是相对偏差。因此,色谱工作站一般都提供两种方法进行峰定性,即:绝对偏差法和相对偏差法。有时候,绝对偏差法也被称为时间带法。这个时候,工作站利用色谱出峰时间的绝对偏差来进行峰定性。前面的例子就是绝对偏差法。绝对偏差法比较灵活,可以单独为每一个色谱峰设置允许偏差,因此被广泛采用。相对偏差法也被称为时间窗法。这个时候,工作站利用色谱出峰时间的相对偏差来进行峰定性。例如,前面的例子设置为允许偏差为2%,则在5.2±2%的范围内的峰,都被认为是乙烷。即从5.1至5.3分钟内的色谱峰,都被认为是乙烷。相对偏差法一般只能设定一个允许偏差值,所有的色谱峰都利用这一个相对偏差来定性,因此灵活性较差。但由于色谱出峰时间越晚,则保留时间变化的可能就越大,而且设置起来方便快捷,因此用相对偏差法来定性也普遍。当采用相对偏差法定性的时候,保留时间小的色谱峰允许偏差很小,因此正常的进样时间误差可能会超过允许偏差,造成定性失误。为此,一些工作站为相对偏差法同时设置了一个小的绝对偏差,例如GC-2010工作站,设置为0.02分钟。在相对偏差之外,另外给了0.02分钟的绝对偏差,确保保留时间小的色谱峰正常定性。例如0.1分钟出峰的色谱峰,2%的允许偏差,自0.08-0.12分钟内的峰,都被认定为该组分。是否有了绝对偏差和相对偏差法,色谱峰就能够被正确定性呢?如果有多个色谱峰进入了允许的偏差范围内,如何确定哪一个色谱峰是特定组分?还是所有色谱峰都是这样一组特定组分?色谱峰的允许误差范围,能不能出现重叠?色谱峰落在重叠范围内的时候,如何确定是哪个特定组分?在没有特定说明,允许偏差范围也没有发生重叠的时候,所有色谱峰都被认为是一个特定组分,或者说是组分群。例如设定碳四组分保留时间为4分钟偏差1分钟,则3-5分钟内的所有色谱峰都被认为是碳四组分。落在两个组分设定偏差的重叠范围内时,通常以前面的峰来定性。例如a组分设定为3.5-4.5分钟,b组分设定为4-5分钟,那么在4-4.5分钟内的组分,会被认定为a组分。即使在3.8分钟已经认定了一个色谱峰为a组分,仍然会把后面的峰认定为a组分。对于复杂样品,组分很多,这样定性就经常会发生问题。因此很多工作站提供更多的功能给峰定性。例如,设置标识峰。在一组保留时间接近的色谱峰中,设置一个标识峰,则认定在此时间范围内面积最大的色谱峰认定为标识峰组分,其他色谱峰则按照设定保留时间与标识峰前后关系进行识别。总之,工作站对于色谱峰的定性,主要有相对和绝对法两种,并采用其他一些技术进行辅助。要正确进行这个工作,需要仔细阅读说明书,并通过实践测试才能得到最好的结果。
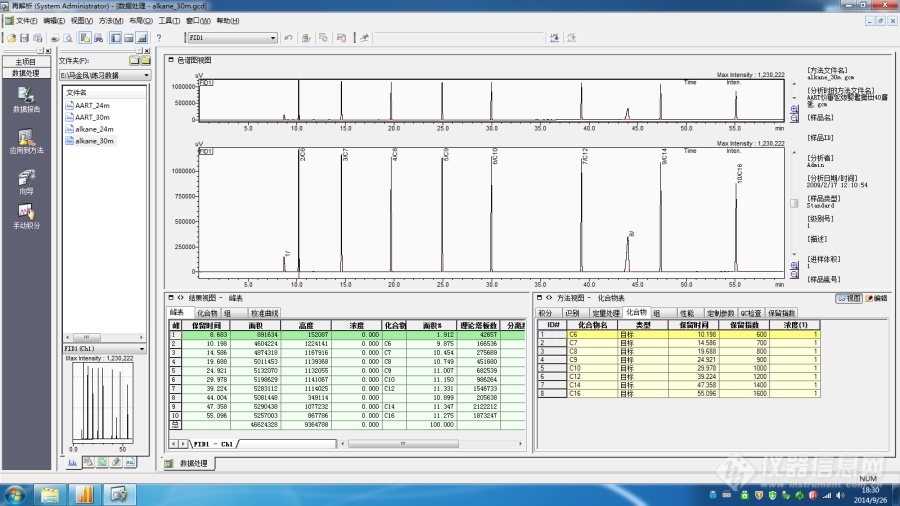
[color=black]Labsolution色谱数据工作站保留指数校正的操作方法[/color][align=center][color=black]概述[/color][/align][color=black]保留指数本质上是规范化的保留时间,在确定的分析条件下,与固定相性质相关,当色谱柱的尺度规格发生变化时,采用保留指数校正功能自动计算和校准定量方法中目标组分的保留时间。[/color][align=center][color=black]一 保留指数校正的原理介绍[/color][/align][color=black]保留指数本质上是规范化的保留时间——是以正构烷烃保留时间为标尺,规范待测组分的保留时间。对于一根确定的色谱柱,在相同的温度和流量操作条件之下,如果色谱柱的具体尺寸规格发生了一定程度的变化,并不影响待测目标物质的保留指数的具体数值。[/color][color=black]例如在农残分析的场合下,色谱工作者在一次进样中可能同时需要处理数十至数百个目标组分的分析。当方法运行一段时间之后,色谱柱需要进行维护。常见的情况下,色谱柱的长度会被明显截短,那么目标组分的保留时间会发生显著的变化。[/color][color=black]此时如果将所有目标组分的保留时间进行重新校准,无疑色谱工作者会面临较大的工作量。如果使用保留指数校正功能的话,只需要重新进样测试一下正构烷烃标准品(前提是方法最初开发时,事先进样过正构烷烃标品),修正分析方法即可,这样可以显著的提高分析效率。[/color][color=black]下文以Labsolution Workstation为例予以说明。[/color][align=center][color=black]二 Labsolution 色谱数据工作站的操作步骤[/color][/align][color=black]假设某项色谱分析,色谱柱原始长度为30m,进样正构烷烃标品获得数据文件为alkane-30.gcd;进样标准样品获得数据文件为AART-30m.gcd。[/color][color=black]当分析进行一段时间之后,色谱柱经过不断维护后长度变成24m。此时进样正构烷烃标品,获得数据文件alkane-24.gcd;进样标准样品获得数据文件为AART-24m.gcd。[/color][color=black]然后基本操作步骤如下:[/color][color=black]2.1 输入原始正构烷烃的保留指数[/color][color=black]进入Labsolution的“再解析”模块,打开alkane-30m.gcd此数据文件,在“方法”界面下,编辑化合物表。此时将正构烷烃的化合物名称、保留时间和保留指数输入到表格中。[/color][color=black]然后保存此数据。[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261803071420_5701_1604036_3.png[/img][/align][color=black]2.2 标准样品的保留指数计算[/color][color=black]打开标准样品数据AART-30m.gcd,然后点击“方法”界面下的“保留指数”选项卡,点击“从数据文件加载”图标,并选择“alkane-30m”此数据文件。[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261803072435_8318_1604036_3.png[/img][/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261803072559_6125_1604036_3.png[/img][/align]然后点击“助手栏”中的“向导”图标,依次点击“下一步”,创建完成方法文件,并保存。此时Labsolution色谱柱数据工作站会自动计算出各个目标组分的保留指数。此时保存方法文件为“标样采集方法”。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261803074199_3900_1604036_3.png[/img][/align]2.3 保留指数的校正当色谱柱长度变为24m后,标样数据中的待测物质保留时间均发生缩短。此时打开柱长24m获得的正构烷烃数据“alkane-24m.gcd”,然后编辑化合物表,填写保留指数,并保存数据问题。然后点击“编辑”菜单,选择“自动修改保留时间AART”。在依次点击“下一步”,此时方法文件中所有目标组分的保留时间,已经同时自动修正为24m下的保留时间。[align=center]小结[/align][color=black]利用保留指数校正功能,可以明显缩短[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法修改消耗的时间,以提升分析效率。[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261803075254_5487_1604036_3.png[/img][/align]
色谱分析定量方法为归一法,分析结果如何以算术平均值报出?
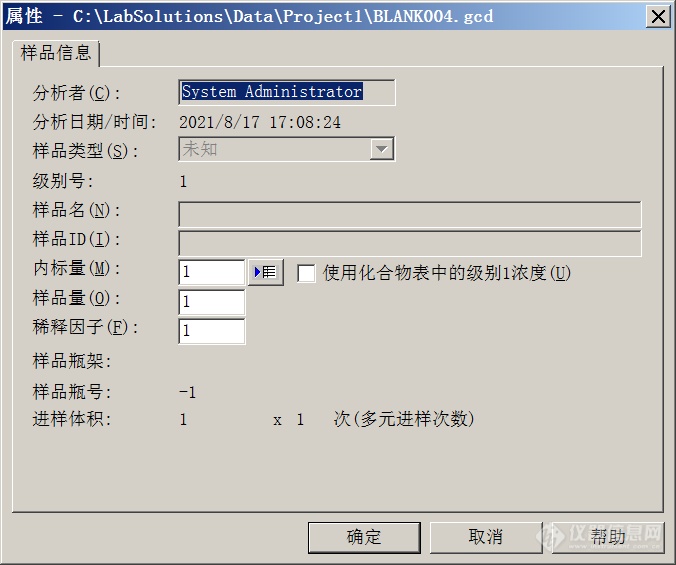
[align=center][font=宋体][font=Calibri]Labsolution[/font][font=宋体]色谱数据工作站定量结果折算的方法介绍[/font][/font][/align]Labsolution色谱数据工作站定量结果折算的方法介绍[align=center]概述[/align]采用Labsolution的“样品量”、“稀释因子”和“定制参数”功能,可以将色谱数据定量结果进行折算。[align=center]背景介绍[/align]在待测样品需要进行前处理操作(例如样品需要溶解、萃取、溶剂提取等),标准样品中待测组分的浓度单位与待测样品中待测组分浓度单位往往会有所不同,色谱分析的结果经常需要进行折算。例如化工分析中固体形态的样品需要称量和溶解;农残分析中待测样品需要称量、粉碎、溶剂提取净化等一系列复杂的操作;当采用内标或者外标法定量时,标准样品的浓度一般会配制为类似mg/l的单位,而原始样品中待测组分的浓度一般需要表示为类似mg/kg的单位。类似上述的情况之下,需要进行不同浓度单位之间的数据折算,色谱工作者一般需要借助第三方的工具或者软件(例如Excel)来实现。其实某些功能较强的色谱数据工作站本身可以完成这项工作,甚至还可以实现更多其他运算功能。下文以Shimadzu 公司的Labsolution Workstation 为例,予以说明,可以利用“稀释因子”、“样品量”和“定制参数”来实现。[align=center] 一 、稀释因子和样品量的使用[/align]以外标法为例,待测样品浓度Ci为:[img=,124,62]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261202195028_4806_1604036_3.png!w124x62.jpg[/img]引入稀释因子和样品量之后,公式则变换为:[img=,214,66]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261202335475_48_1604036_3.png!w214x66.jpg[/img]Labsolution的“分析”模块下,在进样之前的“单次分析开始”界面下,可以根据前处理方法的具体情况,输入合适的样品量和稀释因子。如果数据已经采集完毕,也可以在Labsolution的“再解析”模块下,打开数据,编辑“样品信息”,也可以重新输入样品量和稀释因子。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261201147140_8429_1604036_3.png[/img][/align]也可以在“分析”模块或者“再解析”模块下的“批处理”表中,设定这两个参数。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261201149337_6113_1604036_3.png[/img][/align][align=center]定制参数的使用[/align]色谱工作者如果需要使用更多的计算参数或者计算方法,那就可以使用“定制参数”功能。可以对数据的保留时间、浓度、峰面积、峰高和标准品浓度进行加、减、乘、除四则运算或者乘方开方等运算。可以直接在Labsolution的“再解析”模块下的方法中设定定制参数,已实现此功能。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108261201150568_4637_1604036_3.png[/img][/align]
本文规定了药品注册申报研究工作采用的色谱数据工作站的基本要求和色谱数据的管理要求。同时,为保证色谱数据的完整性和可靠性,色谱数据工作站需建立信息安全管理体系。1、色谱数据工作站基本要求 色谱数据工作站获得的色谱数据应当可靠、安全、完整、可溯源。 鼓励采用经规范和系统验证的色谱数据工作站进行研究工作。 色谱数据工作站验证可由工作站制造商进行,注册申请人依据工作站制造商的评估和验证报告对工作站获得色谱数据的完整性、可靠性、安全性和可溯源性进行评价。色谱数据工作站验证也可由注册申请人自行开展,注册申请人可以通过建立工作站的风险评估办法,制订风险管理文件,对工作站进行评估,确定需要进行验证的项目及内容,并进行系统验证。色谱数据工作站的验证要求将在今后陆续发布。2、色谱数据工作站信息安全管理要求 为保证色谱数据的完整性和可靠性,色谱数据工作站需建立信息安全管理体系。 色谱数据工作站应设系统管理员和信息安全管理负责人。色谱数据只允许经过授权的进入,并能追踪和记录数据的创建、修改和删除。 对于重要色谱数据的任何修改和删除必须获得授权,必须记录修改和删除的原因。重要色谱数据建议采用审计追踪模式记录全部修改和删除情况及原因,审计追踪信息是色谱数据的组成部分,应当和谱图数据和分析结果等仪器归档储存。 色谱数据工作站必须定期对色谱数据进行完全和准确的拷贝。 色谱数据工作站应当可以防止突发情况下色谱数据的丢失,并能追踪和记录到系统的错误和色谱数据错误,同时采取相应的正确措施进行处理。在系统出现故障或瘫痪后,应有明确的和经过验证的恢复处理措施,保证可以将色谱数据恢复到与故障前相同的状态。3、色谱数据的管理要求 色谱数据的存储、保管、存档、备份应当按照本要求进行。 色谱数据的输出需采用符合规定的方式,任何提交的报告的数据应具有可溯源性。3.1 色谱数据的存储、存档和备份 色谱数据应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存,需注意对存储介质的质量、可靠性和耐用性进行评估和选择,注意防止人为或突发情况下色谱数据的丢失和破坏。 应当根据药品研究工作情况构建色谱数据的存档文件(文件夹和命名等)。存档数据应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存,对存储介质的要求同上。 应当定期对色谱数据进行安全备份。备份数据应当保存在独立和安全的设备和存储介质中。对于保存备份数据的存储介质的要求同上。在备份过程中以及备份完成后,应当对备份数据的准确性和完整性进行检查。3.2 色谱数据的输出 用于准备药品注册申报资料的色谱数据的纸面文件应采用色谱数据工作站自动形成的输出文件形式,内容应包括附件1中的相关信息;申报资料的色谱数据的纸面文件还应包括色谱数据的审计追踪信息(如色谱数据的修改删除记录及原因)。 用于纸面存档的色谱数据也应采用色谱数据工作站自动形成的输出文件形式。 不应采用色谱数据工作站软件以外的其他软件进行色谱数据的输出。 不得使用其他软件对色谱数据进行修改。 对于输出的色谱数据,应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存。3.3 色谱数据的保管 色谱数据用于药品注册申报时,在产品获准注册后五年以内所有色谱数据应得到有效保管。 在规定保管期内应定期对存储的色谱数据进行检查,如数据可再次进入情况和数据的准确性。当保管色谱数据的计算机设备或程序发生变化时,必须立即进行检查,确认不会对色谱数据产生影响。3.4数据的可溯源性 任何提交的报告(包括纸面文件)均应可以追踪到相对应的色谱数据。术语色谱数据工作站(workstation of chromatography data) 能完成色谱仪的数据采集、计算、统计、比较、报告、检索、存储功能的装置,还可以具有色谱仪控制、网络支持等扩展功能。色谱数据(chromatography data) 包括仪器信息(仪器编号、仪器控制&序列参数日志等)、样品名称、操作者姓名、谱图数据、分析结果(积分参数&结果、重新积分参数&结果、校准表、报告模板、分析报告等)、审计跟踪信息。验证(validation) 考察证明色谱数据工作站获得的色谱数据是否可靠、安全、完整、可溯源的过程。审计跟踪(audit-trial) 在保证初始的色谱数据不被修改和删除的同时,能够发现和记录对色谱数据的增补、修改、删除详细情况,并能够同时保存这些增补、修改、删除信息。
摘要:目的 建立一种快速测定参枝苓口服液中肉桂酸含量的方法。 方法 首先采用高效液相色谱法测定参枝苓口服液中肉桂酸含量,做为一级数据,并同时采集样品的近红外光谱;采用K-S方法对样品集进行划分考察不同的光谱预处理方法,以Rc、Rv、RMSECV、RMSEP值为模型评价指标。 结果 最佳模型Rc、Rv、RMSECV、RMSEP值分别为0.8862、0.8877、0.00282、0.00301。线性较好,光谱与样品的肉桂酸含量有较好的相关性。结论 建立的参枝苓口服液中肉桂酸含量模型满足生产需求,是一种快速无损、经济的检测方法。关键词:近红外光谱分析技术 参枝苓口服液 肉桂酸 参枝苓口服液是我国首个批准用于老年痴呆的中药复方药物,适用于轻中度阿尔茨海默病所引起的心气虚证,表现症状为心慌心悸、气虚少语、神情冷漠、头晕乏力、失眠健忘、舌淡脉虚等症。组方包括党参、桂枝、茯苓和白芍等10味中药,其中桂枝是主要药材之一,桂枝中主要含有肉桂酸、桂皮醛等成分在药效中起到关键作用。因此,可以通过检测复方中肉桂酸的含量来检验和控制参枝苓口服液的药品质量。目前使用的含量检测方法为HPLC法,该方法比较复杂,不利于实现在线控制。近红外光谱分析技术(Near Infrared Spectroscopy,NIRS)是当前最重要的一种PAT技术,它是指应用波长范围在780-2526nm波长范围内的电磁波的一种快速绿色无损光谱学分析方法并且已经开始广泛应用于农副产品检测、食品检测、药物检测、石油化工产品检测、烟草检测等领域。本实验采用NIRS方法对复方中肉桂酸含量进行测定。1仪器与材料1.1试剂乙腈、甲醇为色谱纯(Merck, Darmstadt, Germany),磷酸为色谱纯(Tedia公司)。肉桂酸对照品购自中国药品生物制品检定所(北京),其余试剂均为分析纯。1.2样品收集四批(生产批号分别为14311、14312、14313、14314)共85个样品,所有样品均有潍坊沃华医药有限公司提供。1.3仪器Agilent 1200高效液相色谱仪,四元泵、DAD检测器、ALS自动进样器、柱温箱Chem-Station for LC 3工作站;柱子型号:Aglient Eclipse Plus C18柱(4.6×250mm,4.6um);Millipore超纯水器(美国Millipore公司);AntarisⅡ傅里叶变换近红外光谱仪(美国Thermo Fisher公司),配有透射模块采样系统和Result操作软件;其他玻璃仪器。光谱处理及模型建立采用Matlab 软件和TQ 软件。2方法2.1近红外光谱的采集采集光谱所用的近红外光谱仪器为美国Thermo Fisher公司生产的AntarisⅡ傅里叶变换近红外光谱仪;将适量样品置于4mm的玻璃样品管中进行光谱采集,光谱采集范围为4000 -10000 cm-1;扫描次数为32;分辨率为8 cm-1;增益值为4x,常温环境采集光谱。每次采集样品前采集背景光谱来消除背景的影响。每个样品的测量时间小于1 min。2.2 HPLC测定芍药苷含量本实验采用HPLC方法作为参考方法来测量样品肉桂酸含量的一级数据。以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%磷酸溶液(25:75)为流动相,待肉桂酸色谱峰出峰后,再用乙腈为流动相洗脱10分钟;检测波长为278nm。理论板数按肉桂酸峰计算应不低于12000。取肉桂酸对照品适量,精密称定,加50%甲醇溶液制成每47.1μg/ml的标准品溶液,即得对照品溶液。精密量取本品10ml,置20ml量瓶中,加甲醇稀释至刻度,摇匀,离心,取上清液,即得供试品溶液。分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,按上述色谱条件,1ml/min流速下等度洗脱50min,记录对应峰面积,外标一点法计算含量。2.3近红外光谱技术分析采用PLS方法将样品液相一级数据与近红外光谱(二级数据)相关联,通过光谱预处理方法的选择、光谱建模区间的选择以及其他建模参数的优化,建立稳健的定量分析模型,并以模型的RMSEC、RMSEP、Rc和Rv值作为模型结果的评价指标。2.3.1校正集和验证集的划分所有的样品划分为校正集和验证集。样品集划分的方法有随机样品划分法(RS),KS法以及SPXY法。在本实验中采用KS法将样品划分为校正集和验证集。KS方法是基于光谱变量的选择方法。根据约4:1的比列将样品划分为68个校正集和17个验证集。2.3.2光谱建模区间的选择本实验分别采用全光谱(4000-10000cm-1)区间以及TQ软件所建议优化的区间(8327-5457cm-1)建立样品的PLS,以Rc、Rv、RMSECV、RMSEP值为参数对模型进行比较,选出最佳的建模光谱区间。2.3.3光谱预处理方法的选择近红外光谱中包含复杂的样品化学信息,在分析过程中,近红外光谱受多种因素的干扰。在建立校正模型之前需要采用化学计量学的方法对光谱作适当的预处理以减少或消除这些影响,改善模型的性能。本实验分别采用SG15、MSC、SNV以及SG15+1d对原始光谱进行预处理并建立PLS数学模型。以Rc、Rv、RMSECV、RMSEP值为参数对模型进行比较,选出最佳的预处理方法。2.3.4模型的建立与模型的评价选用最佳的预处理方法和优化的光谱区间建立PLS模型,用验证集样品对最终模型进行外部验证,并以Rc、Rv、RMSECV、RMSEP值作为模型性能的评价指标。3结果3.1参枝苓口服液肉桂酸含量测定HPLC数据分析表1 参枝苓口服液肉桂酸含量测定统计表 [col
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]系统性试验数据误差的排除方法 使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],在做原料“冰点”含量测定中,发现系统性试验的一组数据136524、173652、165275、193256,其重复性大于2.0。针对这一问题,采取了以下方法进行分析和排除:一、检查进样过程1、注意进样时间的长短,尽量快速进样。2、用校准过的微量注射器进样。3、每次进样操作应当一致,用同样方式和速度进样。二、检查注温是否平稳1、设定检测器温度200℃2、设定进气口温度180℃3、设定柱温80℃4、检查柱箱控温精度在1;柱箱温度波动小于0.1/h;温度梯度波动小于使用温度的2.三、检查柱的老化程度1、检查基线稳定。2、检查谱峰有无拖尾现象。四、检查FTD检测器拆出检测器喷嘴,发现喷嘴内径变小。这是由于固定液流失,样品在喷嘴燃烧后产生积碳,或使用硅烷化衍生物二氧化硅,污染检测器,检测器线性范围变窗,使灵敏度下降。故卸下喷嘴和收集、清洗:1、用通针通喷嘴,必要时用金相砂纸打磨。2、依次用洗洗剂、水超声清洗。3、在100℃-120℃烘干。通过上述方法处理后,再次进行系统性试验,出现另一组数据:163256、163192、163287、1163112、163232,其重复性合理。希望对大家有用!!!
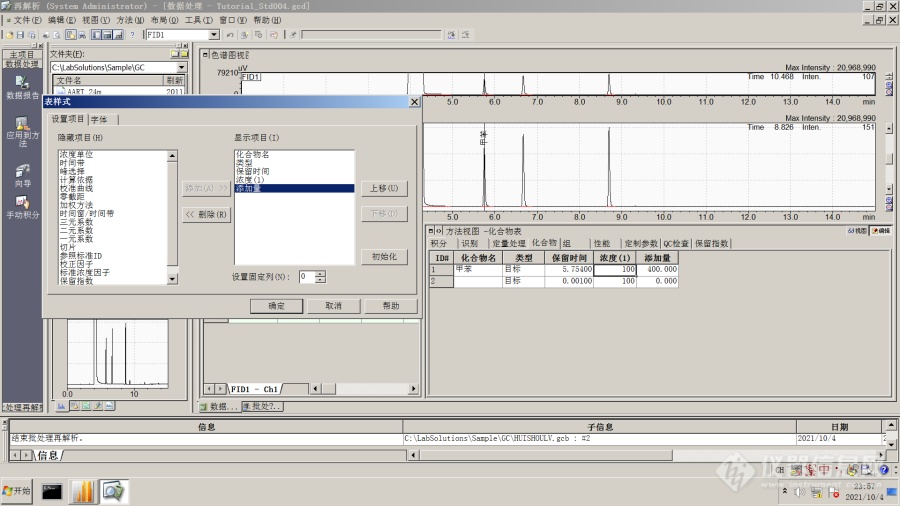
[align=center][size=24px]Labsolutions色谱数据工作站计算回收率的方法[/size][/align][align=center][color=black]概述[/color][/align][color=black]Labsolutions 色谱数据工作站操作教程——回收率的计算。[/color][color=black]在色谱分析过程中,如果待测样品经过某些方式的前处理(例如衍生化、萃取、超声或溶剂提取等),那么就会存在目标物质在前处理过程中是否存在损失的问题,往往需要对前处理方法的准确性作出评价,常用的手段是测定分析方法的回收率。[/color][color=black]常见的实验方式是在样品中添加确定含量的标准物质,然后将样品进行处理之后,测定目标物质的含量,与理论含量的比值为回收率。方法的回收率越高,表示分析方法的准确性越高。 [/color][align=center][color=black]利用Labsolution 色谱数据工作站的QAQC功能计算回收率的操作步骤[/color][/align][color=black]下面以较为简单的单点校准为例进行说明。假设某项色谱分析获得标准品数据为std-01,加标之后的样品用于前处理并测定之后获得的数据为data-01,假设进行加标的待测组分为甲苯,标准品中甲苯含量为100mg/L,样品加标量为400mg/L。[/color][color=black]1 创建分析方法[/color][color=black]在Labsolution的“再解析”部分中,点击“数据处理”模块,打开std-01文件,按照向导创建外标的组分表。[/color][color=black]然后在工作站界面的右下方“方法视图”、“化合物”表中,点击右键,选择“表样式”,增加“添加量”项目。[/color][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110050010276955_1942_1604036_3.png[/img]然后点击“方法”菜单、“QA/QC参数”,编辑回收率选项,注意此时的文件类型为“未知样品”。[img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110050010284244_8991_1604036_3.png[/img]然后保存方法文件,注意保存方法文件时,必须要选中“QA/QC”参数选项。[img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110050010286731_1710_1604036_3.png[/img]2 运行批处理点击Labsolution再解析部分中的“批处理再解析”模块,编写批处理表,将标准品std-01(用来获取标准曲线)和测定回收率样品data-01加入表格。此时注意两个数据文件的文件类型——std-01数据的文件类型为“标准”,data-01数据的文件类型为“未知”。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110050010289029_7043_1604036_3.png[/img][/align]点击“批处理设置”,勾选“执行QAQC”。[img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110050010289250_1603_1604036_3.png[/img]然后保存并且执行此批处理文件,最终可以获得回收率结果,可以用txt文本方式、网页文本方式或者表格方式输出。[img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110050010291812_3608_1604036_3.png[/img]
我在使用浙大N2000气相色谱软件处理数据时,发现同样实验操作做的样品,有的归一化的结果和使用内标法定量的结果相差不足1%,但是有的就能相差23%;比如第一个样品归一化是96.8%,其定量结果是96.1%;第二个样品归一化是93.5%,但是定量可能只有70%左右,这可差距大离谱了,大家有没有碰到这样的问题,欢迎指教。 我测试的样品是乙草胺(自己合成)。 我使用的测试方法进行分流,校正因子又是怎么测试的,究竟怎么样才可以得到准确的乙草胺的纯度数据呢?
气相色谱检测过程中往往会出现假阳性与假阴性的情况,也就是说在复杂基质检测时在目标物质保留时间出现了峰,但不一定就是出现了目标物质,即假阳性;或者色谱图中出现与待测化合物标准品保留时间相近但又不完全一致的色谱峰,那么该如何确定其是杂质还是待测化合物色谱峰,即假阴性;又或者待测化合物有重叠,分离度较低。以上情况我们应该如何分辨,找准目标物质呢,今天就和大家一起来找“李逵”,捉“李鬼”。假阳性如何分辨和避免? 色谱分析中利用保留时间定性是最基本的定性方法,其基本依据是:相同的物质在相同的色谱条件下应该有相同的保留时间。但是,相反的结论却不成立,即在相同色谱条件下,具有相同保留时间的两个色谱峰不一定是同一物质。 分析中经常会碰到复杂基质样品,由于杂质成分与待测化合物性质相似或接近,就可能在色谱行为上表现一致,反应在色谱图中,就是出现与待测化合物标准品保留时间一致的色谱峰,可能是单一组分峰,也可能是多组分重叠峰。对于这些峰,仅靠保留时间定性,我们经常会根据保留时间一致判定为目标化合物,就容易出现误判。出现这种情况,多半是样品未净化完全、待测化合物有基质效应等,尤其在含硫化合物较多的葱、姜、蒜样本的有机磷农药残留检测中经常出现。解决方案检测中,避免出现假阳性结果的方法有:①样品中添加标准品,对比保留时间,并进行加标回收率,看色谱峰及面积是否变化;②改变色谱柱升温程序,看样品中该色谱峰保留时间与标准品保留时间有无差别变化;③利用两根不同极性的色谱柱,或不同类型检测器进行双柱双检测器确证;④采用GC-MS进行确证,利用保留时间和质谱图双重定性,还可结合更严谨的数据分析方法AMDIS进行峰形对照确证,以及二级质谱MS/MS确证;⑤继续优化前处理,进一步去除干扰物后,再分析查看保留时间、峰形等有无变化。假阴性如何分辨和避免? 气相色谱分析中,如果复杂样品基质中某个成分与待测化合物性质相似或接近,就会在色谱出峰上相接近,从而在色谱图中出现与待测化合物对照品保留时间相近的色谱峰。对于这些峰,我们经常会因为保留时间不一致,而定义为杂质峰,不予考虑。但是,实际上仅仅依靠色谱峰保留时间判断,会出现误判,尤其对一些容易出现基质效应的化合物如乙酰甲胺磷、氧化乐果等,这种情况更是经常出现。解决方案 分析中为了避免出现假阴性结果,首先我们可以考虑通过基质标准来定性定量分析,看峰形是否变化,如果有变化,可认为存在基质效应,此时更应该注意;然后考虑调整色谱升温程序(加快或减缓),看能否完全分开;再次,通过双柱双检测器法确证;最后,可利用GC-SIM-MS或GC-MS/MS方法来进一步确证分析,保证结果可靠。如何分辨重叠峰?导致两种化合物tR一致,峰形完全重叠或相近成并肩峰的原因有:①升温程序不合适,温度太高;②载气流速太大;③色谱柱不合适或柱效能太低;④进样量、死体积太大等。如果原来能分开的两色谱峰随着试验进行发生峰重叠而分不开,可能跟以下原因有关:①色谱柱被污染;②色谱柱寿命已到;③载气纯度不佳或净化过滤器失效;④色谱柱温度和载气流量不佳;⑤气化室被污染或样品处理不当,杂质干扰物太多,进样口隔垫漏气;⑥进样技术太差,或进样量超出了色谱柱容量;⑦检测器工作状态变化,如ECD漏气、FID气流比欠佳等;⑧数据处理判峰参数半峰宽或斜率设置不合理,或信号放大器量程、衰减设置失误等。解决方案 针对两个化合物色谱峰重叠的情况,需要逐步分析来解决: 首先考虑是否由进样体积或系统死体积过大导致,可通过减少进样量来进样查看效果; 其次要考虑程序升温速率是否太快,如果是就要将初始温度或升温速率降低,重新进样查看效果,这对于不完全重叠的两个化合物色谱峰,一般都会有所改善,但对完全重叠的两个化合物色谱峰改善效果不大,需进一步改变(降低或增加)载气流速,查看分离情况。 如果上面方式都没有效果,则需要更换不同型号色谱柱或者更高分离性能的色谱柱,提高分离效能。如果仍然无法解决,可考虑进行数据信号模拟处理;或对化合物进行衍生化反应,让其生成新的化合物后再进行测定;或利用质谱仪作为检测器,采用选择离子模式或者二级质谱模式,通过提取化合物的不同电离碎片离子实现分离,增加定性定量结果准确度。

【作者】 雷灼雨; 巴国际;【机构】 重庆市药品检验所; 重庆市药品检验所 重庆 400015; 重庆 400015;【摘要】 目的建立生桂口服液中桂皮醛的含量测定方法。方法采用反相高效液相色谱法,色谱柱为Diamonsil C18柱(150 mm×4.6 mm, 5μm),甲醇-水-冰醋酸(45:55:0.5)为流动相,流速为1.0 mL/min,测定波长为274 nm。结果方法的平均回收率为99.39%,RSD= 0.27%,桂皮醛的线性范围是25.2~201.4μg/mL。结论所建立的方法准确、可靠,能满足该产品的质量控制要求。 更多还原【关键词】 生桂口服液; 桂皮醛; 反相高效液相色谱法; http://ng1.17img.cn/bbsfiles/images/2012/08/201208271555_386456_2352694_3.jpg
标题:高品质的色谱数据工作站------TL 9000金羊CDSTL9000(金羊)新功能功能一:强大的自动检峰能力*智能判峰、基线扣除(new!)、重叠峰校正;*缺省设置,识别所有想要的峰,无论基线、峰分离是否好;功能二:时间程序与手动积分*手动积分处理自动记录到时间程序中(new!);功能三:定量方法百分比法、校正因子归一、内标、外标、指数法、比例因子、多内标(new!)功能四:定性方法手工定性,参比峰定性(new!)、峰表定性(new!),校正表定性,顺序定性法(new!)参比峰定性(new!) 顺序定性法(new!)功能五:校正*校正方式:多点多针校正表一次完成(new!),单点校正,多点校正、手动校正因子功能六:图像处理*图像无极放大缩小(new!)*多种方式的多重绘图与比较(new!)*谱图显示个性化设置(new!)功能七:批次采样存入方法(new!)自动批次采样、处理、存盘、打报告。功能八:快速查找原始数据、报告(new!) 功能九:格式转换*完全灵活的自定义报告格式编辑(new!) *结果可转存为WORD、EXCEL、TXT、MDB,谱图可保存为矢量图EMF联系方式:北京泰立化电子技术有限公司公司网站 WWW.teleh.com.cn联系电话:010-62316613 62316614 62323505 62318086 62318085转300联系人:田明霞手机:13621392881EMAIL:Jane_tmx@126.com
色谱验证方案,原始纪录,验证报告及色谱方法验证实验数据分析[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=112402]色谱验证方案,原始纪录,验证报告及色谱方法验证[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=112404]数据的统计与分析(培训)[/url]
在进行质量研究的过程中,一项重要的工作就是要对质量标准中所涉及到的分析方法进行方法学验证,以保证所用的分析方法确实能够用于在研药品的质量控制。为规范对各种分析方法的验证要求,中国药典2005年版附录规定了分析方法验证的指导原则。该指导原则对需要验证的分析方法及验证的具体指标做了比较详细的阐述。但是文中未涉及各具体指标在验证时的可接受标准,国际上已颁布的指导原则中也未发现相关的要求。另一方面,大多数药品研发单位在进行质量研究时,已逐步认识到分析方法验证的必要性与重要性,大都也在按照指导原则的要求进行分析方法验证,但验证完后却因没有一个明确的可接受标准,而难以判断该分析方法是否符合要求。本文提出了在对HPLC含量测定方法进行验证时的可接受标准,供大家讨论。1.准确度 该指标主要是通过回收率来反映。验证时一般要求分别配制浓度为80%、100%和120%的供试品溶液各三份,分别测定其含量,将实测值与理论值比较,计算回收率。 可接受的标准为:各浓度下的平均回收率均应在98.0%-102.0%之间,9个回收率数据的相对标准差(RSD)应不大于2.0%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为: 在80%至120%的浓度范围内配制5份浓度不同的供试液,分别测定其主峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。 可接受的标准为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。 3.精密度 1)重复性 配制6份相同浓度或分别配制浓度为80%、100%和120%的供试品溶液各三份的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于2.0%。 2)中间精密度 配制6份相同浓度的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于2.0%。 4.专属性 可接受的标准为:空白对照应无干扰,主成分与各有关物质应能完全分离,分离度不得小于2.0。以二极管阵列检测器进行纯度分析时,主峰的纯度因子应大于980。 5.检测限 主峰与噪音峰信号的强度比应不得小于3。 6.定量限 主峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液主峰的保留时间的相对标准差应不大于2.0%。 7.耐用性 分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、流速相对值变化±20%时,仪器色谱行为的变化,选择至少三个不同厂家或不同批号的同类色谱柱,每个条件下各测试两次。可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,分离度应大于1.5;各条件下的含量数据(n=6)的相对标准差应不大于2.0%。 8、系统适应性 配制6份相同浓度的供试品溶液进行分析,主峰峰面积的相对标准差应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,分离度应大于1.5,供试品主峰的理论塔板数应取耐用性试验不同厂家或不同批号的同类色谱柱的平均值的100%-120%。
药审中心发布药品研究色谱数据工作站及色谱数据管理要求(一)本文规定了药品注册申报研究工作采用的色谱数据工作站的基本要求和色谱数据的管理要求。同时,为保证色谱数据的完整性和可靠性,色谱数据工作站需建立信息安全管理体系。1、色谱数据工作站基本要求色谱数据工作站获得的色谱数据应当可靠、安全、完整、可溯源。鼓励采用经规范和系统验证的色谱数据工作站进行研究工作。色谱数据工作站验证可由工作站制造商进行,注册申请人依据工作站制造商的评估和验证报告对工作站获得色谱数据的完整性、可靠性、安全性和可溯源性进行评价。色谱数据工作站验证也可由注册申请人自行开展,注册申请人可以通过建立工作站的风险评估办法,制订风险管理文件,对工作站进行评估,确定需要进行验证的项目及内容,并进行系统验证。色谱数据工作站的验证要求将在今后陆续发布。2、色谱数据工作站信息安全管理要求为保证色谱数据的完整性和可靠性,色谱数据工作站需建立信息安全管理体系。色谱数据工作站应设系统管理员和信息安全管理负责人。色谱数据只允许经过授权的进入,并能追踪和记录数据的创建、修改和删除。对于重要色谱数据的任何修改和删除必须获得授权,必须记录修改和删除的原因。重要色谱数据建议采用审计追踪模式记录全部修改和删除情况及原因,审计追踪信息是色谱数据的组成部分,应当和谱图数据和分析结果等仪器归档储存。色谱数据工作站必须定期对色谱数据进行完全和准确的拷贝。色谱数据工作站应当可以防止突发情况下色谱数据的丢失,并能追踪和记录到系统的错误和色谱数据错误,同时采取相应的正确措施进行处理。在系统出现故障或瘫痪后,应有明确的和经过验证的恢复处理措施,保证可以将色谱数据恢复到与故障前相同的状态。3、色谱数据的管理要求色谱数据的存储、保管、存档、备份应当按照本要求进行。色谱数据的输出需采用符合规定的方式,任何提交的报告的数据应具有可溯源性。 3.1 色谱数据的存储、存档和备份色谱数据应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存,需注意对存储介质的质量、可靠性和耐用性进行评估和选择,注意防止人为或突发情况下色谱数据的丢失和破坏。应当根据药品研究工作情况构建色谱数据的存档文件(文件夹和命名等)。存档数据应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存,对存储介质的要求同上。应当定期对色谱数据进行安全备份。备份数据应当保存在独立和安全的设备和存储介质中。对于保存备份数据的存储介质的要求同上。在备份过程中以及备份完成后,应当对备份数据的准确性和完整性进行检查。
有没有大佬知道,xcalibur软件怎么导出色谱数据啊,跪求!!!
本文规定了药品注册申报研究工作采用的色谱数据工作站的基本要求和色谱数据的管理要求。同时,为保证色谱数据的完整性和可靠性,色谱数据工作站需建立信息安全管理体系。1、色谱数据工作站基本要求 色谱数据工作站获得的色谱数据应当可靠、安全、完整、可溯源。 鼓励采用经规范和系统验证的色谱数据工作站进行研究工作。 色谱数据工作站验证可由工作站制造商进行,注册申请人依据工作站制造商的评估和验证报告对工作站获得色谱数据的完整性、可靠性、安全性和可溯源性进行评价。色谱数据工作站验证也可由注册申请人自行开展,注册申请人可以通过建立工作站的风险评估办法,制订风险管理文件,对工作站进行评估,确定需要进行验证的项目及内容,并进行系统验证。色谱数据工作站的验证要求将在今后陆续发布。2、色谱数据工作站信息安全管理要求 为保证色谱数据的完整性和可靠性,色谱数据工作站需建立信息安全管理体系。 色谱数据工作站应设系统管理员和信息安全管理负责人。色谱数据只允许经过授权的进入,并能追踪和记录数据的创建、修改和删除。 对于重要色谱数据的任何修改和删除必须获得授权,必须记录修改和删除的原因。重要色谱数据建议采用审计追踪模式记录全部修改和删除情况及原因,审计追踪信息是色谱数据的组成部分,应当和谱图数据和分析结果等仪器归档储存。 色谱数据工作站必须定期对色谱数据进行完全和准确的拷贝。 色谱数据工作站应当可以防止突发情况下色谱数据的丢失,并能追踪和记录到系统的错误和色谱数据错误,同时采取相应的正确措施进行处理。在系统出现故障或瘫痪后,应有明确的和经过验证的恢复处理措施,保证可以将色谱数据恢复到与故障前相同的状态。3、色谱数据的管理要求 色谱数据的存储、保管、存档、备份应当按照本要求进行。 色谱数据的输出需采用符合规定的方式,任何提交的报告的数据应具有可溯源性。 3.1 色谱数据的存储、存档和备份 色谱数据应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存,需注意对存储介质的质量、可靠性和耐用性进行评估和选择,注意防止人为或突发情况下色谱数据的丢失和破坏。 应当根据药品研究工作情况构建色谱数据的存档文件(文件夹和命名等)。存档数据应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存,对存储介质的要求同上。 应当定期对色谱数据进行安全备份。备份数据应当保存在独立和安全的设备和存储介质中。对于保存备份数据的存储介质的要求同上。在备份过程中以及备份完成后,应当对备份数据的准确性和完整性进行检查。 3.2 色谱数据的输出 用于准备药品注册申报资料的色谱数据的纸面文件应采用色谱数据工作站自动形成的输出文件形式,内容应包括附件1中的相关信息;申报资料的色谱数据的纸面文件还应包括色谱数据的审计追踪信息(如色谱数据的修改删除记录及原因)。 用于纸面存档的色谱数据也应采用色谱数据工作站自动形成的输出文件形式。 不应采用色谱数据工作站软件以外的其他软件进行色谱数据的输出。 不得使用其他软件对色谱数据进行修改。 对于输出的色谱数据,应当采用适当的存储介质(如光盘、磁带机等电子方式和/或纸面文件等物理方式)进行保存。3.3 色谱数据的保管 色谱数据用于药品注册申报时,在产品获准注册后五年以内所有色谱数据应得到有效保管。 在规定保管期内应定期对存储的色谱数据进行检查,如数据可再次进入情况和数据的准确性。当保管色谱数据的计算机设备或程序发生变化时,必须立即进行检查,确认不会对色谱数据产生影响。3.4数据的可溯源性 任何提交的报告(包括纸面文件)均应可以追踪到相对应的色谱数据。术语色谱数据工作站(workstation of chromatography data) 能完成色谱仪的数据采集、计算、统计、比较、报告、检索、存储功能的装置,还可以具有色谱仪控制、网络支持等扩展功能。色谱数据(chromatography data) 包括仪器信息(仪器编号、仪器控制&序列参数日志等)、样品名称、操作者姓名、谱图数据、分析结果(积分参数&结果、重新积分参数&结果、校准表、报告模板、分析报告等)、审计跟踪信息。验证(validation) 考察证明色谱数据工作站获得的色谱数据是否可靠、安全、完整、可溯源的过程。审计跟踪(audit-trial) 在保证初始的色谱数据不被修改和删除的同时,能够发现和记录对色谱数据的增补、修改、删除详细情况, 并能够同时保存这些增补、修改、删除信息。 附件1 色谱数据输出图谱规范要求1、标明使用的色谱数据工作站,并保留色谱数据工作站固有的色谱图谱头信息,包括:实验者、试验内容、进样时间、运行时间等,进样时间(指injection time)精确到秒,对于软件本身使用 “acquired time”、“作样时间”、“试验时间”等含糊表述的,需说明是否就是进样时间。2、应带有存盘路径的数据文件名。这是原始性、追溯性的关键信息,文件夹和文件名的命名应合理、规范和便于图谱的整理查阅。3、色谱峰参数应有保留时间(保留到小数点后三位)、峰高、峰面积、定量结果、积分标记线、理论板数等。
[color=#444444]本人在做职业卫生方法验证,在做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法试验中,发现低浓度样品代入标准曲线,得出结果与理论值相差几倍,如果标准曲线强制归零后,结果正常,请问各位高手,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法回归曲线可以强制归零吗?多谢了!(我们的仪器是岛津GC2030)[/color]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152864]核分段逆回归集成线性判别分析用于质谱数据分类.caj[/url]摘 要 针对高维小样本质谱数据在构造模型时易产生的过拟合现象!变量间的严重共线性!及结构与性质间的非线性关系,采用了核分段逆回归()特征提取集成线性判别分析()新技术"首先以算法完成质谱数据的非线性特征提取,然后在由新特征矢量张成的低维空间构造样本类别的线性判别函数,负责各样本个体类别的判定"将2方法应用于软饮料的质谱数据分类,结果表明:该方法不仅适应质谱数据与性质间的非线性关系,而且可以更少!解释能力更强的特征变量取得更高的分类精度,并能实现在低维特征空间对数据的解释及可视化。







 我要推广仪器
我要推广仪器
 下载APP
下载APP