[B][center]色谱定性与定量分析方法简介[/center][/B] 在我们日常的检测工作中,色谱的使用频率和使用范围越来越宽,相应的定性与定量的分析方法也越来越重要。此文简单的介绍了色谱定性与定量的方法,并且结合简单的例子,适用于对于色谱接触不久的新手。此原文来自于网络,笔者根据工作的实际经验做了简要的加工。粗陋之处,敬请方家不吝指出。 首先需要说明的是,各种色谱分析方法的定性与定量分析的基本方法都是一样的,不管是薄层色谱、液相色谱、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]或者其它种类的色谱。A.色谱的定性分析1.根据保留时间定性在一定的色谱系统和操作条件下,每种物质都有一定的保留时间,如果在相同色谱条件下,未知物的保留时间与标准物质相同,则可初步认为它们为同一物质。为了提高定性分析的可靠性,还可进一步改变色谱条件(分离柱、流动相、柱温等)或在样品中添加标准物质,如果被测物的保留时间仍然与标准物质一致,则可认为它们为同一物质。此法为色谱定性的基本方法,虽有缺憾,但是使用范围非常之广泛。对于目前常用的工作站而言,允许保留时间的误差默认为5%。2.利用不同的色谱方法定性同一样品可以采用多种检测方法检测,如果待测组分和标准物在不同的检测器上有相同的响应行为,则可初步判断两者是同一种物质。在液相色谱中,还可通过二极管阵列检测器比较两个峰的紫外或可见光谱图。3.保留指数定性 在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中,可以利用文献中的保留指数数据定性。保留指数随温度的变化率还可用来判断化合物的类型,因为不同类型化合物的保留指数随温度的变化率不同。4.柱前或柱后化学反应定性在色谱柱后装T型分流器,将分离后的组分导入官能团试剂反应管,利用官能团的特征反应定性。也可在进样前将被分离化合物与某些特殊反应试剂反应生成新的衍生物,于是,该化合物在色谱图上的出峰位置或峰的大小就会发生变化甚至不被检测。由此得到被测化合物的结构信息。5.与其他仪器联用定性将具有定性能力的分析仪器如质谱(MS)、红外(IR)、[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url](AAS)、原子发射光谱(AES,ICP-AES)等仪器作为色谱仪的检测器即可获得比较准确的定性信息。 需要特别指出的是,以上的定量方法都有一定的局限性,在开发新的分析方法的时候,需要各种分析方法混用,以确保定性的准确,因为这是一切定量工作的基础。B.色谱的定量分析 色谱定量分析的理论基础是待测组分的量与它在色谱图上的峰面积(或峰高)成正比。数据处理软件(工作站)可以给出包括峰高和峰面积在内的多种色谱数据。因为峰高比峰面积更容易受分析条件波动的影响,且峰高标准曲线的线性范围也较峰面积的窄,因此,通常情况是采用峰面积进行定量分析。 具体的定量分析方法有如下几种:1. 校正因子定量 绝对校正因子:单位峰面积所对应的被测物质的浓度(或质量),即样品组分的峰面积与相同条件下该组分标准物质的校正因子相乘,即可得到被测组分的浓度。绝对校正因子受实验条件的影响,定量分析时必须与实际样品在相同条件下测定标准物质的校正因子。 相对校正因子:某物质i与一选择的标准物质S的绝对校正因子之比。即相对校正因子只与检测器类型有关,而与色谱条件无关。 2. 归一化法 归一化法是将所有组分的峰面积分别乘以它们的相对校正因子后求和,即所谓"归一",被测组分X的含量可以用下式求得:采用归一化法进行定量分析的前提条件是样品中所有成分都要能从色谱柱上洗脱下来,并能被检测器检测。归一法主要在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中应用。3. 外标法 直接比较法: 将未知样品中某一物质的峰面积与该物质的标准品的峰面积直接比较进行定量。通常要求标准品的浓度与被测组分浓度接近,以减小定量误差。此法相当于简化的标准曲线法,只不过利用了原点(0,0)和标准物质的一点。定量的精度不如标准曲线法。 标准曲线法: 将被测组分的标准物质配制成不同浓度的标准溶液,经色谱分析后制作一条标准曲线,即物质浓度与其峰面积(或峰高)的关系曲线。根据样品中待测组分的色谱峰面积(或峰高),从标准曲线上查得相应的浓度。标准曲线的斜率与物质的性质和检测器的特性相关,相当于待测组分的校正因子。 4. 内标法 内标法是将已知浓度的标准物质(内标物)加入到未知样品中去,然后比较内标物和被测组分的峰面积,从而确定被测组分的浓度。由于内标物和被测组分处在同一基体中,因此可以消除基体带来的干扰。而且当仪器参数和洗脱条件发生非人为的变化时,内标物和样品组分都会受到同样影响,这样消除了系统误差。当对样品的情况不了解、样品的基体很复杂或不需要测定样品中所有组分时,采用这种方法比较合适。 内标物应满足的要求: 在所给定的色谱条件下具有一定的化学稳定性; 在接近所测定物质的保留时间内洗脱下来; 与两个相邻峰达到基线分离; 物质特有的校正因子应为已知的或者可测定; 与待测组分有相近的浓度和类似的保留行为; 具有较高的纯度。 为了进行大批样品的分析,有时需建立校正曲线。具体操作方法是用待测组分的纯物质配制成不同浓度的标准溶液,然后在等体积的这些标准溶液中分别加入浓度相同的内标物,混合后进行色谱分析。以待测组分的浓度为横坐标,待测组分与内标物峰面积(或峰高)的比率为纵坐标建立标准曲线(或线性方程)。在分析未知样品时,分别加入与绘制标准曲线时同样体积的样品溶液和同样浓度的内标物,用样品与内标物峰面积(或峰高)的比值,在标准曲线上查出被测组分的浓度或用线形方程计算。 5. 标准加入法 标准加入法可以看作是内标法和外标法的结合。具体操作是取等量样品若干份,加入不同浓度的待测组分的标准溶液进行色谱分析,以加入的标准溶液的浓度为横坐标,峰面积为纵坐标绘制工作曲线。样品中待测组分的浓度即为工作曲线在横坐标延长线上的交点到坐标原点的距离。由于待测组分以及加入的标准溶液处在相同的样品基体中,因此,这种方法可以消除基体干扰。但是,由于对每一个样品都要配制三个以上的、含样品溶液和标准溶液的混合溶液,因此,这种方法不适于大批样品的分析。 现在的色谱工作站基本上对于前几种的定量方法都能够自动计算,除了第五种“标准加入法”,现在我简单的举一个例子说明。 例:待测样品检测组分a,现将样品等分为三份,向其中分别添加三个浓度梯度的标准样品,添加之后的浓度与峰面积如下:浓度(ppm)峰面积0.1 98990.3 206150.7 40369利用Excel、miniTab或者其他工具作图如下: 那么待测组分的浓度为x=5080.4/50584=0.1004ppm。
[font=宋体][color=black][back=white]定性方法:用已知保留值定性;根据不同柱温下的保留值定性;根据同系物保留值的规律关系定性;双柱、多柱定性;双检测器定性或利用检测器的选择性定性;利用其他物理方法结合定性,如质谱;利用化学反应或物理吸附作用对样品进行预处理。[/back][/color][/font] [font=宋体][color=black][back=white]定量方法:外标法;内标法;叠加法;归一化法。[/back][/color][/font]
[align=center][b][size=24px]气相色谱仪分析的定性依据及定性方法[/size][/b][/align][color=#000000] [size=18px]气相色谱仪[/size][/color][size=18px]的色谱分析包括色谱定性分析和定量分析。今天为大家浅析气相色谱仪的定性分析依据和定性分析方法,仅供色谱工作者参考交流。 (一)气相色谱仪的定性分析依据:气相色谱主要功能不仅是将混合有机物中的各种成分分离开来,而且还要对结果进行定性及定量分析。所谓定性分析就是确定分离出的各组分是什么有机物质,而定量分析就是确定分离组分的量有多少。色谱在定性分析方面远不如其它的有机物结构鉴定技术,但在定量分析方面则远远优于其它的仪器方法。 有机物进入气相色谱后得到两个重要的测试数据:色谱峰保留值和面积,这样气相色谱可根据这两个数据进行定性定量分析。色谱峰保留值是定性分析的依据,而色谱峰面积则是定量分析的依据。 (二)气相色谱仪定性分析方法:气相色谱的定性分析方法主要有保留值定性法、化学[color=#000000]试剂[/color]定性法和检测器定性法。气相色谱的保留值有保留时间和保留体积两种,现在大多数情况下均用保留时间作为保留值。在相同的仪器操作条件和方法下,相同的有机物应有同样的保留时间,即在同一时间出峰。但必须注意:有同样保留时间的有机物并不一定相同。 气相色谱保留时间定性分析方法就是将有机样品组分的保留时间与已知有机物在相同的仪器和操作条件下保留时间相比较,如果两个数值相同或在实验和仪器容许的误差范围之内,就推定未知物组分可能是已知的比较有机物。但是,因为同一有机物在不同的色谱条件和仪器中保留时间有很大的差别,所以用保留时间值对色谱分离组分进行定性只能给初步的判断,绝对多数情况下还需要用其它方法作进一步的确认。一个最常用的确证方法是将可能的有机物加到有机样品中再进行一次气相色谱仪分析,如果有机样品中确含已知有机物的组分,则相应的色谱峰会增大。这样比较两次色谱图峰值的变化,就可以确定前期初步推断是否正确。[/size]
色谱定性与定量内容提要 本书介绍了色谱分析中常用的定性和定量方法。在定性分析中除介绍了经典的保留值定性的各种方法外,对近年来色谱定性分析中越来越多使用的质谱和红外光谱的谱图解析作了较详细的介绍。在定量分析中除介绍了各种定量分析方法外,对数据处理中的误差分析、定量分析结果的评价和表达方法作了较详细介绍。本书还对色谱定性定量分析常用的积分仪和色谱工作站作了介绍。最后对于保证色谱定性和定量分析结果准确的优良实验室规范作了较详细介绍。本书适于从事色谱分析实验的科技人员阅读。对广大工矿企业及科研院所从事色谱分析的工作人员,大专院校非色谱专业的本科生和研究生,以及有关领导和管理人员是一本有用的参考书[color=#DC143C][color=#FFF8DC][color=#DC143C][color=#00FFFF][color=#DC143C][size=4]第一章 概述 第一节 色谱图的获得 第二节 色谱图中得到的信息 第二章 定性分析 第一节 利用保留值定性 …… 第三章 定量分析 第一节 定量分析基础 …… 第四章 积分仪和色谱工作站 第一节 概述 …… 第五章 优良实验室规范和实验室认可制度 第一节 实验室的组织和管理 …… 主要参考文献 附录 附录一 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]检定规程(JJG 700-1990) 附录二 液相色谱仪检定规程(JJG 705-1990)[/size][/color][/color][/color][/color][/color]这里是连接:[URL=http://www.namipan.com/d/%e8%89%b2%e8%b0%b1%e5%ae%9a%e6%80%a7%e4%b8%8e%e5%ae%9a%e9%87%8f.pdf/94a0622e30afe6ac688ef259585749293ded1e6b25c9c800]http://www.namipan.com/d/%e8%89%b2%e8%b0%b1%e5%ae%9a%e6%80%a7%e4%b8%8e%e5%ae%9a%e9%87%8f.pdf/94a0622e30afe6ac688ef259585749293ded1e6b25c9c800[/URL]
本人想系统学习[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]定性与定量方面知识,求助各位高手,小弟在此表示忠心感谢!
色谱定性与定量,如何翻译呢?
[color=#444444]本人实验内容如下:一种固态物质经过物理方法进行预处理后得到固液两项的混合物,希望通过[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]色谱联用,定性定量的测定预处理前后物质中的有机物。请教内否实现,查过许多国内文献少有这方面的研究,通常均是对某一定性物质进行分析检测。[/color]
请问各位专家,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱分析农药残留做定性定量分析时,怎么选择定性和定量离子?依据是什么?谁能分享一下相关的资料!谢谢!
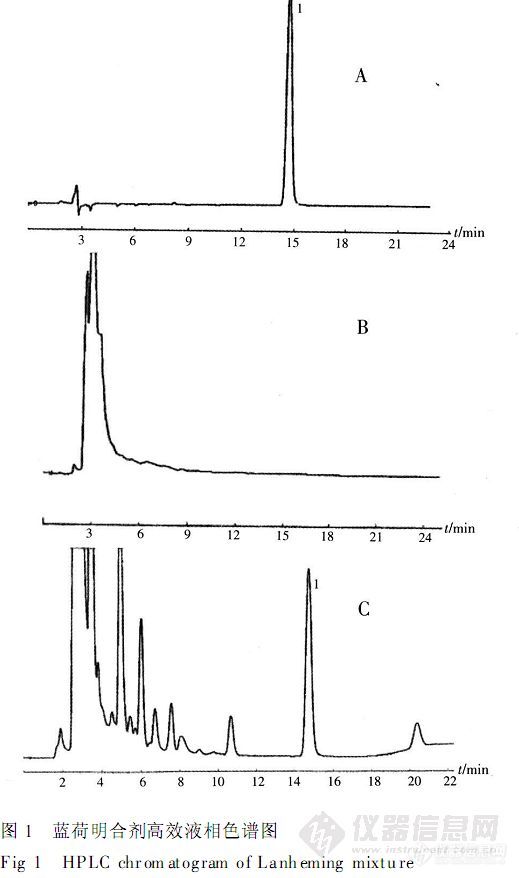
【作者中文名】卜振军; 朱志宏; 徐霞; 阳海;【作者英文名】PU Zhen-jun; ZHU Zhi-hong; XU Xia; YANG Hai(Hunan Jiudian Pharmaceutical Co.; Ltd; Changsha 410011);【作者单位】湖南九典制药有限公司;【摘要】目的建立蓝荷明合剂定性定量方法。方法采用薄层色谱法对蓝荷明合剂中所含荷叶、绞股蓝、决明子进行定性鉴别研究。采用高效液相色谱法测定大黄酚的含量。色谱柱Diamonsil C18(4.6 mm×250 mm,5μm);流动相为甲醇-0.1%磷酸(85∶15);检测波长为254 nm;流速为1.0 mL.min-1。结果薄层色谱鉴别方法专属性强、斑点分离较好。大黄酚平均回收率为98.7%,RSD=0.83%(n=6)。结论所建立的方法可作为蓝荷明合剂的质量控制方法。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061223_381828_2379123_3.jpg
石榴皮中没食子酸定性定量方法研究王克英 (贵州省药品检验所,贵州贵阳550004) 目的:建立石榴皮中没食子酸定性定量的方法。方法:用薄层色谱法对石榴皮有效成分没食子酸进行定性分析,采用高效液相色谱法测定了没食子酸含量。以Diamomil C18柱为固定相,流动相0.2%甲醇乙腈.0.1%磷酸和0.1%三乙胺水溶液(3:9r7),检测波长216 nm,柱温40℃,结果:没食子酸在0.085~O.768螺峰面积与进样量呈良好的线性,r=0.999 6;平均回收率96.7%;RSD 0.8%。结论:通过20批不同产地石榴皮的鉴别和含量测定,证明方法灵敏准确可靠,重复性好。[关键词】石榴皮;没食子酸;TIC;HPIEPS:该文献没有谱图
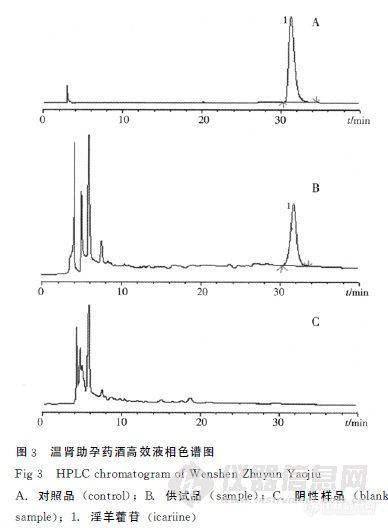
作者:项菲;(怀化市食品药品检验所;)摘要:目的建立温肾助孕药酒的定性定量研究方法。方法用薄层色谱(TLC)法对当归、黄芪进行定性鉴别;用高效液相色谱(HPLC)法测定温肾助孕药酒中淫羊藿苷的含量,采用Dikma Diamonsil C18柱(250mm×4.6mm,5μm);流动相:乙腈-水(30∶70);流速:1.0mL.min-1;柱温:40℃;检测波长:270nm;进样量:10μL。结果 TLC法能明显检出当归、黄芪特征斑点,阴性药材无干扰。淫羊霍苷进样量在0.187 4~1.874μg与峰面积线性关系良好,r=0.999 88,平均回收率为98.6%,RSD=0.88%。结论该定性定量方法简便、准确、重现性好,能有效控制温肾助孕药酒的内在质量。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061029_381708_1606903_3.jpg
液相色谱如何进行定性和定量分析?
定性与定量基础 1 定性定性要解决的问题——在一套色谱图中,目标色谱峰是何种物质。(例如:1号色谱峰是何种物质,或者目标物质是几号色谱峰。) 1 2 3 定性的原则——保留时间(Rt)是定性的依据。 定性的方法——利用标样(含有的全部物质是已知的,或者使用纯品)。 例如:(1)进样标样,已知 1 、2号峰为水、乙醇。 1 2 Rts2=1.0min (2)进样样品,得到谱图如下 1 2 3 Rt2=1.05min 2号峰保留时间Rt2接近标样中乙醇的保留时间Rts2,可认为2号峰即为乙醇。 保留时间(Rt)是定性的依据 2 定量定量要解决的问题——目标物质有多少(含量有多高)。定量的原则——峰面积(或者峰高)是定量的依据。 基本关系式:m = f * A m------- 待测物质的量 f -------- 校正因子 A-------- 待测物质峰面积 定量的方法——利用标样(含有的目标物质是已知的,并且其含量是已知的)。 例如:(单点外标法定量)(1)进样标样,已知 1 、2号峰为水、乙醇。[/s

请问这样的色谱图影响定性与定量结果吗?这个大鼓包可能的原因是什么?谢谢!http://ng1.17img.cn/bbsfiles/images/2017/10/201010162153078128_01_1765567_3.jpg
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]还能用信噪比定性和定量吗?
请问您在没有质谱情况下,用过双色谱柱定性定量吗?
色谱定性、定量分析的基础知识(朋友赠与,共同分享)[color=#DC143C]大家有需要给楼主发站内短信,个人信息在这里留下容易造成安全的隐患 ---- 版主水中月[/color]
求《[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]定性与定量》汪正范(第二版),论坛上一直找不到第二版,有小伙伴能分享以下吗?
各位大神,请问有[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]与质谱联用检测鼠药的方法吗?我查了一下,没找到鼠药的检测标准,查文献也是用液相质谱联用检测的比较多,只求定性方法,有定量的更佳,多谢各位大神解答。
[url=http://www.instrument.com.cn/download/shtml/021616.shtml]色谱定性与定量(电子书)[/url] 希望对大家有帮助!![em61]
各位友人大家好: 我刚开始接触气相色谱质谱联用仪,质谱是四级杆质谱,主要用于定量分析,请问各位能否分享一下:气质定量定性方法开发流程的相关ppt等资料,不胜感激!
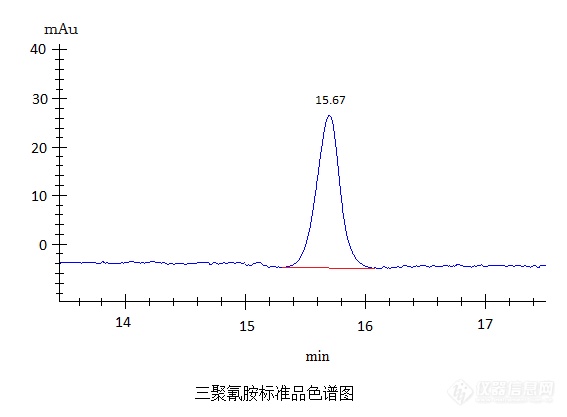
[align=center][font='宋体'][size=24px]色谱定性分析问题[/size][/font][font='宋体'][size=24px]应对方法[/size][/font][font='宋体'][size=24px]你应该知道[/size][/font][/align][font='宋体'][size=16px] 色谱分析大部分都是要定量结果,一小部分只要求定性就可以(只看样品中含不含这种组分,对定量并未做具体要求)。要求定量的大家往往把定量看的很重,定性并不太在意,没有定性[/size][/font][font='宋体'][size=16px]哪来定量,定性都不准谈何定量(定性[/size][/font][font='宋体'][size=16px]分析是确定样品中有无我们要检测的物质,该物质通过那些色谱数据体现,色谱定性分析大多是靠相同条件下标准品物质保留[/size][/font][font='宋体'][size=16px]时间来确定,相同物质不管在样品中还是标准品中保留时间基本一致。定量分析是通过计算定性分析确定的物质的色谱峰面积或峰高与标准品色谱峰面积或峰高的关系[/size][/font][font='宋体'][size=16px]。说白了定性分析是通过某种方法确定样品中是否有目标物质,是色谱分析的第一步[/size][/font][font='宋体'][size=16px];[/size][/font][font='宋体'][size=16px]定量分析是通过某种方法计算样品中目标物质的含量[/size][/font][font='宋体'][size=16px]。[/size][/font][font='宋体'][size=16px])[/size][/font][font='宋体'][size=16px] 色谱定性分析非常重要,要想分析准确,定性分析必不可少。下面我们就具体介绍介绍高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析中常见一些定性分析的问题。[/size][/font][font='宋体'][size=16px] 有时候比如中药材、乳制品、生物制品、肉制品等中某些物质的特性很接近,色谱峰保留时间很接近[/size][/font][font='宋体'][size=16px],通过定性分析很难确定目标物质。这在高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析中就很难办了,不能准确找到目标物质,目标物质的含量也就不能准确检测、计算出来。[/size][/font][font='宋体'][size=16px] 以乳制品中三聚氰胺高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]检测为例。[/size][/font][font='宋体'][size=16px]由于三聚氰胺和某种蛋白质性质很接近[/size][/font][font='宋体'][size=16px](在以前有人甚至把三聚氰胺称作是“蛋白精”),定性分析也是这个检测的一个难点。比如我们在检测中碰到的一些情况。[/size][/font][font='宋体'][size=16px]三聚氰胺标准品保留时间[/size][/font][font='宋体'][size=16px]15[/size][/font][font='宋体'][size=16px].67min[/size][/font][font='宋体'][size=16px],样品一有这种物质保留时间与标准品保留时间较接近,分别是[/size][/font][font='宋体'][size=16px]15[/size][/font][font='宋体'][size=16px].[/size][/font][font='宋体'][size=16px]3[/size][/font][font='宋体'][size=16px]1min[/size][/font][font='宋体'][size=16px],[/size][/font][font='宋体'][size=16px]15[/size][/font][font='宋体'][size=16px].[/size][/font][font='宋体'][size=16px]6[/size][/font][font='宋体'][size=16px]5min[/size][/font][font='宋体'][size=16px];样品二两种分别是[/size][/font][font='宋体'][size=16px]15[/size][/font][font='宋体'][size=16px].[/size][/font][font='宋体'][size=16px]5[/size][/font][font='宋体'][size=16px]2min[/size][/font][font='宋体'][size=16px],[/size][/font][font='宋体'][size=16px]15[/size][/font][font='宋体'][size=16px].[/size][/font][font='宋体'][size=16px]9[/size][/font][font='宋体'][size=16px]4min[/size][/font][font='宋体'][size=16px];样品三两种分别是[/size][/font][font='宋体'][size=16px]15[/size][/font][font='宋体'][size=16px].72min[/size][/font][font='宋体'][size=16px],[/size][/font][font='宋体'][size=16px]16[/size][/font][font='宋体'][size=16px].[/size][/font][font='宋体'][size=16px]1[/size][/font][font='宋体'][size=16px]9min[/size][/font][font='宋体'][size=16px]。[/size][/font][font='宋体'][size=16px]样品中目标物质与标准品物质保留时间前后相差零点[/size][/font][font='宋体'][size=16px]几[/size][/font][font='宋体'][size=16px]分钟是很正常的,像以上这三个样品[/size][/font][font='宋体'][size=16px]中[/size][/font][font='宋体'][size=16px]哪一个是三聚氰胺[/size][/font][font='宋体'][size=16px]就很难确定。[/size][/font][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210021406379836_3016_2369266_3.png[/img][font='宋体'][size=16px] 下面我们就介绍一种实验员常用的也是比较有效的方法[/size][/font][font='宋体'][size=16px]—[/size][/font][font='宋体'][size=16px]给样品中加入一定量标准品,通常是假设色谱峰高较低的[/size][/font][font='宋体'][size=16px]那个为目标物质加的量[/size][/font][font='宋体'][size=16px]尽量大一点,一般[/size][/font][font='宋体'][size=16px]是那个色谱峰算出来的含量的[/size][/font][font='宋体'][size=16px]1[/size][/font][font='宋体'][size=16px]倍以上[/size][/font][font='宋体'][size=16px]的量[/size][/font][font='宋体'][size=16px](量小了不容易看出来),重新进样。加标后样品,[/size][/font][font='宋体'][size=16px]如果假设的那个色谱峰比原来高了,高的量大概和理论的量差不多(原来色谱峰高的几倍,倍数和加标量接近),另外临近的[/size][/font][font='宋体'][size=16px]那[/size][/font][font='宋体'][size=16px]个[/size][/font][font='宋体'][size=16px]色谱峰高无明显变化,那说明我们假设的那个就是目标物质。反之假设的这个色谱峰高无明显变化,另外临近的那个色谱峰高有明显变化,那说明另外那个是目标物质。[/size][/font][img=,690,259]https://ng1.17img.cn/bbsfiles/images/2022/10/202210071220297263_1012_2369266_3.png!w690x259.jpg[/img][img=,690,296]https://ng1.17img.cn/bbsfiles/images/2022/10/202210071906376633_9732_2369266_3.png!w690x296.jpg[/img][font='宋体'][size=16px] 这种方法使用有一定条件。一是样品或标准品保留时间必须稳定,如果相邻的几次进样同一物质保留时间变化较大,那这种方法用起来可能也会出错。二是[/size][/font][font='宋体'][size=16px]多次进样色谱图类似度较高,[/size][/font][font='宋体'][size=16px]在相同的时间区间[/size][/font][font='宋体'][size=16px]不能一次进样出两个峰,一次进样出一个或多个峰,[/size][/font][font='宋体'][size=16px]那这种方法很难分辨出来。三[/size][/font][font='宋体'][size=16px]如果是两种组分[/size][/font][font='宋体'][size=16px]色谱图上体现出来的必须是两个色谱峰,如果两种[/size][/font][font='宋体'][size=16px]组分[/size][/font][font='宋体'][size=16px]只出了一个色谱峰(保留时间较近,两色谱峰完成重叠到一块或是大的色谱峰[/size][/font][font='宋体'][size=16px]覆盖住了小的色谱峰[/size][/font][font='宋体'][size=16px])[/size][/font][font='宋体'][size=16px],我们很可能就会认为该色谱峰就是目标物质出的峰,也就不会耗时费力的去做确认等其它工作了。[/size][/font][font='宋体'][size=16px] 遇到以上这种较难的定性分析的情况[/size][/font][font='宋体'][size=16px],我们首先得有个决策,看看能不能确认那个是目标物质色谱峰,如果确认不了,看看加标这种方法适不适用,适用了我们就用不适用了我们在想其它方法。[/size][/font][font='宋体'][size=16px] 这种色谱定性分析方法较常见也较适用,色谱定性分析这一类问题如何应对,应对方法你应该知道。[/size][/font]
关于质谱图中的定量离子与定性离子的确定方法:定量离子是不是以质谱图中最高的相对离子强度为定量离子,定性离子的选择则以特征离子的丰度比为选择依据啊,我是初学者,多谢指教
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=96252]实验室认证《色谱技术丛书——色谱定性与定量(第二版)》.[/url]
一、定性分析(qualitative spectrometric analysis)定性分析∶ 就是对研究对象进行"质"的方面的分析。定性依据∶元素不同→电子结构不同→光谱不同→特征光谱1、元素的灵敏线灵敏线∶最易激发的能级所产生的谱线,每种元素都有一条或几条谱线最强的线,即灵敏线。最后线也是最灵敏线;元素特征光谱中强度最大的谱线为元素的灵敏线。在供试品光谱中,应检出某元素的灵敏线,从而确定是否存在待测元素。2、定性方法3、标准光谱比较定性法为什么选铁谱?(1)谱线多∶ 在210~660nm范围内有数千条谱线 (2)谱线间距离分配均匀∶容易对比,适用面广 (3)定位准确∶ 已准确测量了铁谱每一条谱线的波长。标准谱图∶将其他元素的分析线标记在铁谱上,铁谱起到标尺的作用。谱线检查∶将试样与纯铁在完全相同条件下摄谱,将两谱片在映谱器(放大器)上对齐、放大20倍,检查待测元素的分析线是否存在,并与标准谱图对比确定。可同时进行多元素测定。二、定量分析 (spectrometric analysis)1、光谱半定量分析与目视比色法相似 测量试样中元素的大致浓度范围;应用∶ 用于钢材、合金等的分类、矿石品位分级等大批量试样的快速测定。谱线强度比较法∶ 测定一系列不同含量的待测元素标准光谱系列,在完全相同条件下(同时摄谱),测定试样中待测元素光谱,选择灵敏线,比较标准谱图与试样谱图中灵敏线的黑度,确定含量范围。2、光谱定量分析(1) 发射光谱定量分析的基本关系式在条件一定时,谱线强度Ⅰ 与待测元素含量c关系为∶I= a ca为常数(与蒸发、激发过程等有关),考虑到发射光谱中存在着自吸现象,需要引入自吸常数 b,则∶[img=,181,73]https://pic2.zhimg.com/80/v2-7043df0c3917bff02f9772eb0991a2f5_1440w.webp[/img]发射光谱分析的基本关系式,称为塞伯-罗马金公式(经验式)。自吸常数 b随浓度c增加而减小,当浓度很小,自吸消失时,b=1。(2) 内标法基本关系式影响谱线强度因素较多,直接测定谱线绝对强度计算难以获得准确结果,实际工作多采用内标法(相对强度法)。│在被测元素的光谱中选择一条作为分析线(强度I),再选择内标物的一条谱线(强度I0),组成分析线对。则∶相对强度RA为其他三项合并后的常数项,内标法定量的基本关系式。内标元素与分析线对的选择∶a.内标元素可以选择基体元素,或另外加入,含量固定;b.内标元素与待测元素具有相近的蒸发特性;c.分析线对应匹配,同为原子线或离子线,且激发电位相近(谱线靠近),"匀称线对";d.强度相差不大,无相邻谱线干扰,无自吸或自吸小。
新人刚接触[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],请教大神,1.[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]能对一未知有机混合物准确定性吗?请简单介绍一下。2.FID检测器测含ABC三种成份的未知有机物,用哪种方法能准确测出三种的含量?3.两台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测同一混合物质中某组份,相同条件测试下会有多大误差?比较啰嗦请大神不吝赐教,万分感谢。
[align=center]高效液相色谱组分定性分析方法简述[/align]1、保留时间对照法(1)外标对照法 即在相同的色谱条件下,分别进样品溶液与高纯度的单一组分对照品溶液,这里推荐先进样品溶液,确定系统适应性没有问题了再进对照品溶液,对比两组分的保留时间,一般保留时间相对差异在5%以内,同时绝对误差在0.1min以内的认定为同一个物质,但仍遵守“同一组分保留时间肯定一样,但保留时间一样不一定是同一组分,保留时间不一样的肯定不是同一组分”的原则。(2)标准加入法 即在相同的色谱条件下,将高纯度的对照品加入样品溶液中再上机检测,与相同浓度未加对照品的样品溶液比较,峰高或峰面积应呈等比例增加的,可认定为是同一物质。此法比较适用于组分比较复杂,邻近有干扰组分峰时。上述方法尽量使用柱效高或长柱,峰高尽量小一些,甚至可以小到定量限浓度,一些在高响应下是单个峰的,在低响应时可能是两个峰,所以一定要注意峰形,只要峰形与对照品不一致,就要怀疑不是同一个物质。再严谨一点,还可以改变流动相组成、色谱柱、柱温等,样品中的组分峰应与对照品的组分峰有相同的变化。2、利用检测器选择性进行鉴别(1)DAD检测器波长扫描法 对于配有DAD检测器的还可以对比三维扫描图谱和峰纯度计算进行辅助定性。如果没有配DAD检测器,但有具波长扫描功能的可变波长检测器的,可以使用停泵扫描功能,查看扫描图谱进行鉴别。(2)质谱检测器鉴别法 该方法适用于联用有质谱分析仪的高效液相色谱仪,通过比较碎片峰进行比较鉴别。3、破坏法 将物质进行化学破坏,可以是酸、碱降解,也可以是衍生后再进行检测,看组分峰的变化。当然,此方法不适用于组分复杂的样品。
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]定性和定量分析基本原理
面积归一法、内标法、外标法、标准加入法。这么多定量法,用哪一种呢?每一种的适用范围是什么呢?其优缺点又是什么?其标准物又有什么要求呢?看完你就都知道了。归一化法把所有出峰的组分含量之和按100%计的定量方法,称为归一化法。各成分校正因子一致时可用该法,该法简便、准确,特别是进样量不容易准确控制时,进样浓度及进样量的变化的影响很小。其他操作条件,如流速、柱温等变化对定量结果的影响也很小。GC应用广于HPLC。外标法(标准曲线法、直接比较法)首先用欲测组分的标准样品绘制标准工作曲线。具体作法是:用标准样品配制成不同浓度的标准系列,在与欲测组分相同的色谱条件下,等体积准确量进样,测量各峰的峰面积或峰高,用峰面积或峰高对样品浓度绘制标准工作曲线,此标准工作曲线应是通过原点的直线。若标准工作曲线不通过原点,说明测定方法存在系统误差。标准工作曲线的斜率即为绝对校正因子。当欲测组分含量变化不大,并已知这一组分的大概含量时,也可以不必绘制标准工作曲线,而用单点校正法,即直接比较法定量。单点校正法实际上是利用原点作为标准工作曲线上的另一个点。因此,当方法存在系统误差时(即标准工作曲线不通过原点),单点校正法的误差较大。因此规定,y=ax+b 。b的绝对值应不大于100%响应值是y的2%。标准曲线法的优点:绘制好标准工作曲线后测定工作就很简单了,计算时可直接从标准工作曲线上读出含量,这对大量样品分析十分合适。特别是标准工作曲线绘制后可以使用一段时间,在此段时间内可经常用一个标准样品对标准工作曲线进行单点校正,以确定该标准工作曲线是否还可使用。标准曲线法的缺点:每次样品分析的色谱条件(检测器的响应性能,柱温度,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。另外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),因此对样品前处理过程中欲测组分的变化无法进行补偿。内标法选择适宜的物质作为欲测组分的参比物,定量加到样品中去,依据欲测组分和参比物在检测器上的响应值(峰面积或峰高)之比和参比物加入的量进行定量分析的方法称为内标法。内标法的关键是选择合适的内标物。内标物应是原样品中不存在的纯物质,该物质的性质应尽可能与欲测组分相近,不与被测样品起化学反应,同时要能完全溶于被测样品中。内标物的峰应尽可能接近欲测组分的峰,或位于几个欲测组分的峰中间,但必须与样品中的所有峰不重叠,即完全分开。一般会选择标准物质的同位素物质作为内标物。内标法的优点:进样量的变化,色谱条件的微小变化对内标法定量结果的影响不大,特别是在样品前处理(如浓缩、萃取,衍生化等)前加入内标物,然后再进行前处理时,可部分补偿欲测组分在样品前处理时的损失。若要获得很高精度的结果时,可以加入数种内标物,以提高定量分析的精度。内标法的缺点:选择合适的内标物比较困难,内标物的称量要准确,操作较麻烦。使用内标法定量时要测量欲测组分和内标物的两个峰的峰面积(或峰高),根据误差叠加原理,内标法定量的误差中,由于峰面积测量引起的误差是标准曲线法定量的2-2,但是由于进样量的变化和色谱条件变化引起的误差,内标法比标准曲线法要小很多,所以总的来说,内标法定量比标准曲线法定量的准确度和精密度都要好。标准加入法标准加入法实质上是一种特殊的内标法,是在选择不到合适的内标物时,以欲测组分的纯物质为内标物,加入到待测样品中,然后在相同的色谱条件下,测定加入欲测组分纯物质前后欲测组分的峰面积(或峰高),从而计算欲测组分在样品中的含量的方法。标准加入法的优点:不需要另外的标准物质作内标物,只需欲测组分的纯物质,进样量不必十分准确,操作简单。若在样品的前处理之前就加入已知准确量的欲测组分,则可以完全补偿欲测组分在前处理过程中的损失,是色谱分析中较常用的定量分析方法。标准加入法的缺点:要求加入欲测组分前后两次色谱测定的色谱条件完全相同,以保证两次测定时的校正因子完全相等,否则将引起分析测定的误差。
所谓多元是指多个波长处的谱带,单元是指单个波长处的谱带。在红外光谱中(不是近红外)人们似乎多用单元线性回归和最多两元的比例线性回归来进行定量分析。我很想知道,像近红外那样用多元线性分析方法来分析红外光谱,是不是一个用效的定性(如种类的鉴别)和定量(如组分的比例)分析方法?







 我要推广仪器
我要推广仪器
 下载APP
下载APP