[align=center][b][size=24px]气相色谱仪四种定量方法 [/size][/b] [/align] 1、面积内标法 取标准被测成分,按依次增加或减少的已知阶段量,各自分别加入各单体所规定的定量内标准物质中,调制标准溶液。分别取此标准液的一定量注入气相色谱仪色谱柱,根据气相色谱仪上色谱图取标准被测成分的峰面积和峰高和内标物质的峰面积和峰高的比例为纵座标,取标准被测成分量和内标物质量之比,或标准被测成分量为横坐标,制成标准曲线。然后按单体中所规定的方法调制试样液。在调制试样液时,预先加入与调制标准液时等量的内标物质。然后按制作标准曲线时的同样条件下得出的色谱,求出被测成分的峰面积或峰高和内标物质的峰积或峰高之比,再按标准曲线求出被测成分的含量。 2、面积外标法 取标准样品成分,在测标准样品之前就算出所取标准样品中含有成分的量,再用气相色谱法测得标准样品的峰面积,然后去标准被测物质,气相色谱法测该物质的峰面积,两者峰面积相比较,最后得出含量值。所用的外标物质,应采用其峰面积的位置与被测成分的峰的位置尽可能接近并与被测成分以外的峰位置完全分离的稳定的物质即标样,一般使用99.5%纯度的气相色谱仪色谱专用化学试剂样品。这也是目前大多数气相色谱仪建议采用的检测方法。 3、绝对标准曲线法 取标准被测成分按依次增加或减少阶段法,各自调制成标准液,注入一定量后,按色谱图取标准被测成分的峰面积或峰高为纵座标,而以标准被测成分的含量为横坐标,制成标准曲线。然后按单体中所规定的方法制备试样液。取试样液按制标准曲线时相同的条件作出色谱,求出被测成分的峰面积和峰高,再按标准曲线求出被测成分的含量。 4、峰面积百分率法 以色谱中所得各种成分的峰面积的总和为100,按各成分的峰面积总和之比,求出各成分的组成比率。根据色谱上出现的物质成分的峰面积或峰高进行定量。峰面积可用面积测定仪测定,按半宽度法求得(即以峰1/2处的峰宽×峰高求得)。峰高的测定方法是从峰高的顶点向记录纸横座标准垂线,找出此垂线与峰的两下端联结线的交点,即以此交点至峰顶点的距离长度为峰高。
硬度计的分类有很多,具体有以下几种分类方法: 一、硬度计按材料分类可分为: 金属硬度计、橡胶硬度计、石材硬度计、水果硬度计。 二、硬度计按名称分类有: 洛氏硬度计 HR 表面洛氏硬度计 HR(15.30.45)(N,T) 维氏硬度计 HV 里氏硬度计 HL 布氏硬度计 HB(S) 肖氏硬度计 HS 邵氏硬度计 HA 韦氏硬度计 HW 巴氏硬度计 一个单位相当于压入深度0.0076mm 三、硬度计按类型分类有: 便携硬度计(手提式硬度计)、台式硬度计、多功能硬度计、超声波硬度计、视觉硬度计。 四、硬度计按示值显示分类有: 数显硬度计、指针示硬度计、读值查表硬度计。来源:卓腾网
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]常用的四种类型
顶空+气相色谱有四种牌子,岛津,PE,安捷伦,戴安,各牌子比较新的型号都有哪些?其详细参数在哪里能找到?
简化二级核共振氢谱的常用四种方法是什么?这是我们明天就要交的作业,哪位好心人能指教一下?平时没听现在不会做____-------[img]http://simg.instrument.com.cn/bbs/images/brow/em09506.gif[/img]
谁有SN/T 3788-2014 进出口纺织品中四种有机氯农药的测定 气相色谱法 这个标准共享下,
请问,有机氟化物的烯烃(含异构体),环氧化物,酮。四种物质。有什么适合的色谱柱吗?
高效液相色谱-串联质谱法测定樱桃番茄中四种季铵类农药张曦 金芬 钱永忠 于志勇 王静 【摘要】:建立了4种季铵类农药(矮壮素、燕麦枯、敌草快和缩节胺)的高效液相色谱-串联质谱(LC-MS/MS)检测方法。样品用75%乙腈-水溶液提取,高速离心后取上清液过尼龙滤膜,用亲水柱分离,以含0.1%甲酸的水:乙腈=25:75(V:V)作为流动相,在正离子多反应监测(MRM)模式下进行测定。4种季铵类农药的线性范围为0.5~100ng/mL,线性相关系数在0.9982~0.9996范围内,方法检出限为0.005mg/kg,可以满足国际限量的要求。在0.04、0.08、0.16mg/kg添加浓度下,4种季铵类农药的回收率为91.4%~106.3%,相对标准偏差(RSD)小于15%。本方法可用于樱桃番茄样品中4种季铵类农药的测定。【作者单位】: 中国农业科学院农业质量标准与检测技术研究所农产品质量与食物安全重点实验室;中国科学院生态环境研究中心国家水质学重点实验室; 【关键词】: 季铵化合物 农药 液相色谱-串联质谱法 樱桃番茄 【基金】:中央级公益性科研院所基本科研业务费专项(0032007228) 2008年农业行业标准制修订项目资助 【分类号】:TS255.7【正文快照】: 季铵类化合物作为除草剂和植物生长调节剂在农业上有着广泛的应用,常见的季铵类农药包括燕麦枯(Difenzoquat,DF)、敌草快(Diquat,DQ)、矮壮素(Chlormequat,CQ)和缩节胺(Mepiquat,MQ)等(结构见图1)。敌草快对生物体的氧化还原活性影响很大,如肺、心、肝、肾等均有不同程度的伤
归一化法把所有出峰的组分含量之和按100%计的定量方法,称为归一化法。各成分校正因子一致时可用该法,该法简便、准确,特别是进样量不容易准确控制时,进样浓度及进样量的变化的影响很小。其他操作条件,如流速、柱温等变化对定量结果的影响也很小。GC应用广于HPLC。外标法(标准曲线法、直接比较法)首先用欲测组分的标准样品绘制标准工作曲线。具体作法是:用标准样品配制成不同浓度的标准系列,在与欲测组分相同的色谱条件下,等体积准确量进样,测量各峰的峰面积或峰高,用峰面积或峰高对样品浓度绘制标准工作曲线,此标准工作曲线应是通过原点的直线。若标准工作曲线不通过原点,说明测定方法存在系统误差。标准工作曲线的斜率即为绝对校正因子。当欲测组分含量变化不大,并已知这一组分的大概含量时,也可以不必绘制标准工作曲线,而用单点校正法,即直接比较法定量。单点校正法实际上是利用原点作为标准工作曲线上的另一个点。因此,当方法存在系统误差时(即标准工作曲线不通过原点),单点校正法的误差较大。因此规定,y=ax+b b的绝对值应不大于100%响应值是y的2%。标准曲线法的优点:绘制好标准工作曲线后测定工作就很简单了,计算时可直接从标准工作曲线上读出含量,这对大量样品分析十分合适。特别是标准工作曲线绘制后可以使用一段时间,在此段时间内可经常用一个标准样品对标准工作曲线进行单点校正,以确定该标准工作曲线是否还可使用。标准曲线法的缺点:每次样品分析的色谱条件(检测器的响应性能,柱温度,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。另外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),因此对样品前处理过程中欲测组分的变化无法进行补偿。内标法选择适宜的物质作为欲测组分的参比物,定量加到样品中去,依据欲测组分和参比物在检测器上的响应值(峰面积或峰高)之比和参比物加入的量进行定量分析的方法称为内标法。内标法的关键是选择合适的内标物。内标物应是原样品中不存在的纯物质,该物质的性质应尽可能与欲测组分相近,不与被测样品起化学反应,同时要能完全溶于被测样品中。内标物的峰应尽可能接近欲测组分的峰,或位于几个欲测组分的峰中间,但必须与样品中的所有峰不重叠,即完全分开。内标法的优点:进样量的变化,色谱条件的微小变化对内标法定量结果的影响不大,特别是在样品前处理(如浓缩、萃取,衍生化等)前加入内标物,然后再进行前处理时,可部分补偿欲测组分在样品前处理时的损失。若要获得很高精度的结果时,可以加入数种内标物,以提高定量分析的精度。内标法的缺点:选择合适的内标物比较困难,内标物的称量要准确,操作较麻烦。使用内标法定量时要测量欲测组分和内标物的两个峰的峰面积(或峰高),根据误差叠加原理,内标法定量的误差中,由于峰面积测量引起的误差是标准曲线法定量的2-2是由于进样量的变化和色谱条件变化引起的误差,内标法比标准曲线法要小很多,所以总的来说,内标法定量比标准曲线法定量的准确度和精密度都要好。标准加入法标准加入法实质上是一种特殊的内标法,是在选择不到合适的内标物时,以欲测组分的纯物质为内标物,加入到待测样品中,然后在相同的色谱条件下,测定加入欲测组分纯物质前后欲测组分的峰面积(或峰高),从而计算欲测组分在样品中的含量的方法。标准加入法的优点:不需要另外的标准物质作内标物,只需欲测组分的纯物质,进样量不必十分准确,操作简单。若在样品的前处理之前就加入已知准确量的欲测组分,则可以完全补偿欲测组分在前处理过程中的损失,是色谱分析中较常用的定量分析方法。标准加入法的缺点:要求加入欲测组分前后两次色谱测定的色谱条件完全相同,以保证两次测定时的校正因子完全相等,否则将引起分析测定的误差。(来源:互联网)
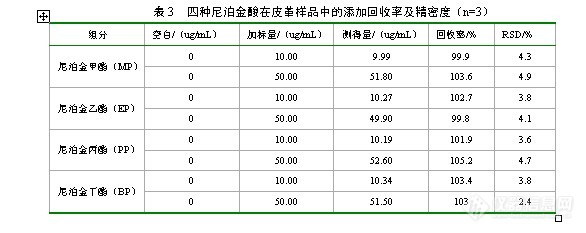
高效液相色谱法检测食品中四种尼泊金酯类防腐剂摘要建立了一种用高效液相色谱法快速检测食品中四种尼泊金酯类防腐剂的分析方法。提取液经Agilent Eclipse XDB-C18液相色谱柱(250x 4.6mm,5 um)分离,流动相为甲醇-0.02mol/L乙酸铵(45:55)进行梯度洗脱,柱温箱温度为30℃,采用可变波长紫外检测器检测,检测波长254nm。四种尼泊金酯类防腐剂在(0.5~200)ug/mL范围内线性关系良好,相关系数均大于0.995,检出限为(0.032~0.046) mg/L(S/N=3),加标回收率为(99.8~105.2)% ,RSD小于5%。该方法操作简便、快捷、灵敏、准确,适合食品中尼泊金酯类防腐剂的定量测定。关键词高效液相色谱法;食品;尼泊金酯类;防腐剂High Performance Liquid Chromatography Detection of Four Kinds of Food Parabens PreservativesAbstract: Food for the rapid detection of a high-performance liquid chromatography analysis of the four kinds of parabens preservatives. Extract Agilent Eclipse XDB-C18 HPLC Columns (250x 4.6mm, 5 um) column, the mobile phase gradient elution of methanol-0.02mol / L ammonium acetate (45:55), a variable wavelength UV detector detected, the detection wavelength of 254nm. Four kinds of parabens preservatives (0.5 to 200) ug / mL linearity in the range of a good relationship, the correlation coefficient greater than 0.995, the detection limit (0.032 to 0.046) mg / L (S / N = 3) recoveries (99.8 ~ 105.2)%, RSD less than 5%. The method is simple, fast, sensitive, accurate and suitable for leather products, parabens preservatives quantitative determination.Key words:high performance liquid chromatography;food; Parabens; preservatives1 前言尼泊金酯也称对羟基苯甲酸酯,是国际上公认的广谱、高效、低毒类防腐剂。在食品、化妆品、纺织品与皮革和医药等领域中广泛使用。尼泊金酯具有酚羟基结构,抗细菌性能比苯甲酸、山梨酸都强
请问各位大侠啦,四种六六六的质谱图的碎片离子是一样的吗?[em0815]
出现圆头峰有四种可能进样量过大 检测器污染灵敏度下降载气系统存在泄露
ROHS中新加入的四种物质有一段时间了,不知道大家都在用什么方法测,有出报告吗?
GC-MS进α、β、γ、δ四种六六六标样,结果色谱图上出了五个峰,质谱显示有两个峰均为α-六六六,二者保留时间相差有2分钟左右,不知何故,大家日常工作中遇见过这种情况吗?
[color=#444444]用液相色谱分离四种植物激素,峰形分分离度不够,如果调整流动相梯度,请问应该怎么调整?原理是什么?原则是什么?[/color][color=#444444]附件里有我的色谱图形和流动相梯度,谢谢。[/color][color=#444444][img=,690,314]https://ng1.17img.cn/bbsfiles/images/2019/07/201907191118359293_6246_1676638_3.jpg!w690x314.jpg[/img][/color]
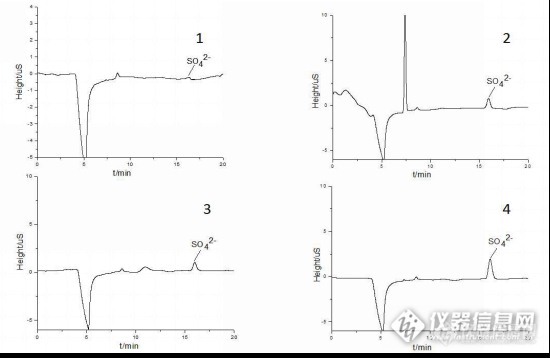
在线固相萃取-离子色谱法测定四种芳环磺酸盐中的硫酸根离子摘要:建立一种在线固相萃取离子色谱法测定四种芳环磺酸盐中硫酸根离子含量的新方法,将自装填的PGC-SPE柱应用于离子色谱系统对样品进行在线前处理,样品经过PGC-SPE柱处理后进入500μL定量环,通过阀切换-大体积进样模式使硫酸根进入阴离子检测系统。固相萃取流路以1.5mmol/L Na2CO3在0.8mL/min的流速对基体在线富集;分析柱采用SH-AC-3 (4.0×250mm) + SH-AG-3 (4.0×50mm),在6mmol/L Na2CO3 +4mmol/L NaHCO3条件下等度洗脱,柱温为35 ℃,流速为0.8mL/min,进样量20μL。结果表明:硫酸根离子在0.50-10.00 mg/L浓度范围内呈良好的线性关系,线性相关系数为0.9993,保留时间、峰高、峰面积的RSD均在0.28%~2.86%之间,方法检出限为0.0106mg/L,回收率在93.33%~105.59%之间;该方法具有良好的线性和重复性,整个在线分析过程在25min之内完成,进样量少,快速、高效。关键字:在线固相萃取,离子色谱,多孔石墨化碳,自装填技术,硫酸根离子1 前言多孔石墨化碳(PGC)作为一种新型色谱固定相,因其表面为纯碳结构、平整且无接枝的功能基团而具有耐强酸强碱、耐高温、性能稳定的特点,广泛应用于液相色谱和离子色谱中。PGC是二维结构、表面完全非极性且有可自由移动的π电子,因此与它化合物之间存在着疏水作用和电子间作用等多重作用力,从而在一定条件下对极性化合物、非极性化合物、异构体、聚合物等都具有保留性且对强极性化合物表现出强保留性。在样品前处理特别是复杂样品前处理过程中,通常利用固相萃取对样品进行净化与浓缩富集,除去干扰性的杂质,从而提高待测物分离度并保护色谱柱。Thermo公司首先推出以多孔石墨化碳填料为固定相的hypercarb 分析柱。在固相萃取技术应用中已有商品化的离线多孔石墨化碳固相萃取小柱(PGC-SPE柱),主要应用于液相色谱和离子色谱的样品的富集和基体消除。 萘磺酸盐和蒽醌磺酸盐属于多苯环磺酸盐,易溶于水的强极性离子型化合物。目前商品化的离子色谱固相萃取前处理柱,如:Onguard RP柱、H柱、Na柱以及聚合物树脂固相萃取柱等对非离子型有机化合物和常规离子基质有很好的前处理效果,但对芳环磺酸盐类的离子型强极性有机化合物保留性差,前处理效果不理想;在测定这类物质中残留的无机离子时,若不能很好地对该类物质进行基体消除,其进入分析系统后易保留在离子色谱柱上且较难洗脱,对分析柱造成损害。PGC因其特殊的表面疏水性质和带电性质使得它对该类强极性离子型化合物具有很强的保留性。本实验创新性的提出自装填可再生的多孔石墨化碳固相萃取柱并将其应用于在线离子色谱分析系统对四种芳环磺酸盐中的硫酸根离子进行测定,自装填和可再生重复使用的特点很大程度上降低了实验成本,在线基体消除较离线前处理在能保证理想的前处理效果和样品分析重复性的同时,能很大程度的减少样品污染并缩短了分析时间和样品量,更有利于复杂样品中阴离子的快速、准确的分离分析。2 实验部分1.1 仪器与试剂实验所用仪器为自组装离子色谱系统,主要部件包括:Ultimate3000 WPS-3000TSL自动进样器,ICS5000+紫外检测器,ICS5000+(SP/DP)双泵,ICS5000 TCC柱温箱(含一个六通阀和一个十通阀),ICS3000(SP)单泵,ED50A电化学检测器(DS3,带控温)Chromeleon6.8色谱工作站,(美国赛默飞世尔科技有限公司);WLK-6A阴离子抑制器(青岛仪趣仪器有限公司);AL-10电子天平(梅特勒-托利多有限公司);Milli-Q Advantage A10超纯水机(Millipore);BRANSON 2510超声清洗仪(BRANSON); 硫酸根离子标准储备液(100 mg/L,上海计量测试技术研究院);碳酸钠和碳酸氢钠(分析纯,99%,上海凌峰化学试剂有限公司);氢氧化钠 (w/w=50% ,默克公司);浓磷酸(分析纯,国药集团化学试剂有限公司);甲基磺酸(99.5%,上海阿拉丁有限公司);甲醇,乙腈(色谱纯,上海星可高纯溶剂有限公司)。 1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾(上海翰思化工有限公司提供)1.2 标准溶液的配制 分别取0.50,1.00,2.00,5.00,10.00 mL的SO42-标准储备液于100 mL容量瓶,超纯水定容后得到0.50,1.00,2.00,5.00,10.00 mg/L的SO42-标准液。 准确称取0.0242g、0.0213g、0.0248g、0.0257g的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾于50mL容量瓶,超纯水溶解定容,作为待测样品。1.3 色谱条件在线PGC-SPE柱为本实验室自装填前处理小柱(3.0×30mm),PGC填料粒径:30μm,阴离子分析柱: SH-AC-3 (4.0×250mm,9μm) + SH-AG-3 (4.0×50mm,12μm) (青岛盛翰色谱技术有限公司),柱温:35 ℃,流速:0.8mL/min;进样量20μL;定量环:500μL;淋洗液:6mmol/LNa2CO3 +4mmol/LNaHCO3; 分析时间25min;1.4 On-line SPE-IC系统的构建 On-line PGC-SPE-IC系统构建如图1 所示,实验建立了阀切换-大体积进样抑制电导离子色谱方法测定四种芳环磺酸盐中的硫酸根离子。系统中泵1用于在线固相萃取和进样,泵2用于阴离子分析,泵3用于SPE柱的在线清洗再生;利用一个十通阀和一个六通阀实现样品的在线固相萃取、大体积进样和在线前处理柱的再生,抑制电导检测器对硫酸根离子进行检测。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131609_609526_3137073_3.jpg 图1 在线SPE-IC系统2 结果与讨论2.1 PGC-SPE柱的装填和活化及柱容量 实验中所使用的SPE柱为本实验室自装填前处理小柱,采用湿法装填方法,以乙醇为分散剂,将填料以流动相的形式利用高压泵进行装填,装填过程先是填料在低流速低压条件下缓慢泵入柱体内,一段时间后再缓慢将流速调节为1mL/min装填1h以上,最后加大流速在高压条件下将填料压实;装填好的SPE柱压力约为120 psi。将装填好的PGC-SPE柱进行一下酸碱活化过程:酸冲洗(100mmol/LHCl, 1mL/min,30min)——水冲洗(1mL/min,20min)——碱冲洗(100mmol/LNaOH, 1mL/min,30min)) —— 水洗(1mL/min,20min)。酸碱冲洗的作用在于对小柱进行活化同时溶解填料中本身残留的一些酸碱可溶性杂质,避免其进入色谱系统损害色谱柱和抑制器。 利用紫外检测基线突越的方法测定PGC-SPE柱对该四种化合物的柱容量。分别配置500mL浓度为230.0、236.4、129.0、258.0mg/L的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾溶液作为流动相,以0.1mL/min的流速流过SPE柱,记录基线开始采集和发生突越的时间间隔,根据公式计算(柱容量=物质浓度*流速*时间)得:1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾的柱容量分别是0.8912mg,0.8307mg,0.2354mg和0.4373mg。2.2 色谱分析条件的选择2.2.1 硫酸根在SPE柱上洗脱条件(泵1淋洗液) 在中性条件下, 由于电荷的相互作用,PGC-SPE柱对硫酸根具一定的保留性,而酸性或者碱性条件下将其洗脱;实验中考虑到仪器系统兼容性和耐碱程度,选择低浓度碳酸钠作为洗脱液。由于使用500uL定量环大体积进样方式,流速为0.8mL/min,则SO42-的峰展宽必须控制在0.625min之内。根据实验结果可知(见图2),在0.5mM,1mM,1.5mM碳酸钠条件下,阴离子完全洗脱时间分别是1.10-2.20min,0.75-1.45min,0.65-1.20min;1.5mM浓度下的硫酸根离子在0.55min之内洗脱,因此Pump1选择1.5mM碳酸钠作为洗脱液。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131601_609519_3137073_3.jpg 图

【作者】 张雪; 褚文静; 刘伟娜; 侯海妮; 黄喜茹;【Author】 ZHANG Xue,CHU Wen-jing,LIU Wei-na,HOU Hai-ni,HUANG Xi-ru(Department of Analytical Chemistry,The School of Pharmaceutical Sciences,Hebei Medical University,Shijiazhuang 050017)【机构】 河北医科大学药学院分析化学教研室;【摘要】 建立了高效液相色谱二极管阵列检测(HPLC-DAD)法同时测定丹参滴注液中丹参素、原儿茶醛、迷迭香酸和丹酚酸B四种水溶性成分的含量。采用DiamonsilTMC18色谱柱(250×4.6 mm,5μm),以甲醇和5%冰乙酸为流动相进行梯度洗脱,流速为1.0mL/min,柱温30℃,检测波长为286 nm。在此色谱条件下四种水溶性成分可完全分离。丹参素、原儿茶醛、迷迭香酸和丹酚酸B的线性范围分别为0.2192~1.934μg(r=0.9999),0.03508~0.2456μg(r=1.0000),0.2592~1.814μg(r=1.0000),0.3864~2.705μg(r=0.9999)。平均回收率丹参素为102.6%,相对标准偏差(RSD)为0.55%;原儿茶醛为103.5%,RSD为0.42%;迷迭香酸为99.8%,RSD为0.68%;丹酚酸B为102.8%,RSD为0.49%。该方法简单、快速,四组分分离良好,可用于丹参滴注液的质量控制。http://ng1.17img.cn/bbsfiles/images/2012/07/201207181222_378455_1761902_3.jpg
请问哪位老师做过鸡肉中四种沙星同时检测啊,麻烦您把出峰顺序和时间或者谱图告诉我,谢谢~
[align=center][b][size=16px]实验室需要掌握的四种比对方法[/size][/b][/align][size=15px]化学分析计量[/size] [size=15px][color=var(--weui-FG-2)]2022-12-28 20:00[/color][/size] [size=15px][color=rgba(0, 0, 0, 0)]发表于山东[/color]今天给大家总结实验室需要掌握的四种内部比对方法:人员比对、方法比对、仪器比对、留样再测。一)人员比对[/size] [size=16px] 人员比对试验是指在相同的环境条件下,采用相同的检测方法、相同的检测设备和设施,由不同的检测人员对同一样品进行检测的试验。 当某项试验可由多人进行操作时,实验室可采用人员比对试验的方式进行内部质量控制,通过安排具体具有代表性的不同层次的两人或者多人展开,考核测试人员的能力水平,判断检测人员操作是否正确、熟练、用以评价人员对试验检测结果准确性、稳定性和可靠性的影响。作为实验室内部质量控制的手段、人员比对优先适用于以下情况:1)依靠检测人员主观判断较多的项目、例如食品中的感官、品尝的项目;2)在培员工和新上岗的员工;3)检测过程的关键控制点或关键控制环节;4)操作难度大的项目和或者样品;5)检测结果在临界值附近;6)新安装的设备;7)新开验的检测项目。二)方法比对 方法比对试验是指在环境条件相同、相同的人员采用不同的检测方法对同一样品进行的检测。当某个检测项目可以由多种方法进行操作时,实验室可以采用方法比对进行内部质量控制,判断检测所遵循的标准或者方法是否被严格地理解和执行,用以评价检测方法对试验检测结果准确性、稳定性和可靠性的影响。作为实验室内部质量控制的手段、方法比对优先适用于以下情况:1)刚实施的新标准或者新方法;2)引进的新技术、新方法和研制的新方法;3)已有的具有多个检验标准或方法的项目。三)仪器比对 仪器比对试验是指在相同的环境、相同的方法、由相同的检测人员采用不同的仪器设备对同一样品进行检测的试验。 当某项试验可由多种设备进行操作时,实验室可采用设备比对试验的方式进行内部质量控制,判断对测量准确度、有效性有影响的设备是否符合测量溯源性的要求,用以评价仪器设备对实验室检测结果准确性、稳定性和可靠性的影响。作为内部质量控制手段,设备比对试验优先适用于以下情况:①新安装的设备;②修复后的设备;③检测结果出现在临界值附近的设备。四)留样再测 留样再测是指在尽可能相同的环境条件下,采用相同的检测方法、相同的检测设备和设施,由相同的检测人员对已完成检测的样品在其留样保存期间进行再次检测的试验。 实验室通过留存样品的再次测量,比较分析上次测试结果与本次测试结果的差异,用以发现实验室因偶然因素对实验室检测结果准确性、稳定性和可靠性的影响。 作为内部质量控制手段,留样再测可在下列情况采用:①验证检测结果的准确性;②验证检测结果的重复性;③对留存样品特性的监控。[/size]
成分分析中四种以上的成分大家采用的是那个方法?怎么来减少误差?
根据光谱干扰产生的特点,光谱干扰分四种类型,如图所示。 ? 1)简单平滑光谱背景分析线谱峰被平滑背景叠加后,平行向上移动。采用离峰单点校正法,可以准确地扣除光谱背景引起的干扰。即在谱峰一侧或两侧测出背景强度值,从含有背景值的峰值强度中扣除之。 2)斜坡背景这类光谱背景随波长渐变的,但其变化是线性的。用离峰左右两点法可以对其进行准确校正。即在谱峰两侧等距离地测定两处背景强度,然后取其平均值从峰值强度的测量值中扣除之。 3)弯曲背景分析线处在共存元素高强度谱峰的一翼,形成渐变弯曲斜坡背景。如果分析线强度较大,则仍可按线性斜坡背景的扣除法进行校正。但如果分析线强度较低,则这种校正背景方法误差太大,会给出错误的分析数据。为了准确校正弯曲斜坡背景,要用多个离峰测点准确地描绘光谱背景,然后扣除背景值。对于这种光谱背景的校正,用在峰法或差谱法是很简便的。即用不含待测元素的溶液(含有等量形成光谱干扰的元素)在峰测出背景值,然后从样品中扣除之。 4)复杂结构背景及谱线重叠这种光谱背景通常由分子谱带或谱线混合叠加形成。对于这种背景,采用空白溶液校正法是比较有效的。简单的离峰单点或两点法校正往往得出错误的结果,单单根据一幅样品溶液的谱图无法得知峰谱下光谱背景的真实情况。[img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106191549397982_7250_2140715_3.png[/img]
美国药典中有对铅 镉 砷 汞四种重金属的检测 么 如有分别是按什么方法检测的啊 着急!!
色谱法的基本原理利用样品混合物中各组分理、化性质的差异,各组分程度不同的分配到互不相溶的两相中。当两相相对运动时,各组分在两相中反复多次重新分配,结果使混合物得到分离。两相中,固定不动的一相称固定相;移动的一相称流动相。分类:根据流动相分—以气体作流动相—[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]——固定相为液体 气-液色谱 固定相为固体 气-固色谱 —以液体作流动相—液相色谱——固定相为液体 液-液色谱 固定相为固体 液-固色谱 —当流动相是在接近它的临界温度和压力下工作的液体时——超临界色谱 根据固定相的附着方式 —固定相装在圆柱管中—柱色谱 —固定相涂敷在玻璃或金属板上—薄膜色谱(平板色谱) —液体固定相涂在纸上—纸色谱(平板色谱)根据分离机理 —分配色谱—样品组分的分配系数不同 —吸附色谱— 样品组分对固定相表面吸附力不同 —体积排阻色谱—利用固定相孔径不同,把样品组分按分子大小分开 —离子交换色谱—不同离子与固定相商相反电荷间的作用力大小不同根据极性 —流动相极性>固定相极性-反相色谱 —流动相极性<固定相极性-正相色谱 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]只适合分析较易挥发、且化学性质稳定的有机化合物,而HPLC则适合于分析那些用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]难以分析的物质,如挥发性差、极性强、具有生物活性、热稳定性差的物质。所以,HPLC的应用范围已经远远超过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]。 一、吸附色谱(adsorption chromatography)又叫液固色谱法:流动相是液体,固定相是固体。分离原理:固定相是固体吸附剂,吸附剂是多孔性微粒物质表面有吸附中心。样品组分与流动相竞争吸附中心。各组分的吸附能力不同,使组分在固定相中产生保留时间不同和实现分离。固定相: 固定相通常是强极性的硅胶、氧化铝、活性炭、聚乙烯、聚酰胺等固体吸附剂。活性硅胶最常用。流动相: 弱极性有机溶剂或非极性溶剂与极性溶剂的混合物,如正构烷烃(己烷、戊烷、庚烷等)、二氯甲烷/甲醇、乙酸乙酯/乙腈等。 应用: 对于极性,结构异构体分离和族分离仍是最有效的方法,如农药异构体分离、石油中烷、烯、芳烃的分离。 缺点是容易产生不对称峰和拖尾现象。二、分配色谱原理: 固定液机械的吸附在惰性载体上,样品分子依据他们在流动相和固定相间的溶解度不同,分别进入两相分配而实现分离。固定相:将一种极性或非极性固定液吸附在惰性固相载体上。如全多孔微粒硅胶吸附剂。根据极性不同分类:正相分配色谱—固定相载体上涂布的是极性固定液; 流动相是非极性溶剂; 可分立极性较强的水溶性样品; 弱极性组分先洗脱出来。 反相分配色谱—固定相载体上涂布的是非极性或弱极性固定液; 流动相是极性溶剂; 强极性组分先洗脱出来。 液-液分配色谱固定相中的固定液体往往容易溶解到流动相中去,所以重现性很差,且不能进行梯度洗脱,已经不大为人们所采用。三、键合相色谱 考虑分配色谱法中固定液的缺点,因此将各种不同的有机关能团通过化学反应共价结合到固定相惰性载体上,固定相就不会溶解到流动相中去了。键合固定相优点:○ 对极性有机溶剂有良好的化学稳定性 ○使色谱柱的柱效高、寿命长 ○实验重现性好 ○几乎适于各种类相的有机化合物的分离,尤其是k’宽范围的样品 ○可以梯度洗脱根据极性不同分类:正相键合相色谱—固定相极性>流动相极性 固定相:二醇基、醚基、氰基、氨基等极性基团的有机分子。 适于分离脂荣、水溶性的极性、强极性化合物 反相键合相色谱—固定相极性<流动相极性 固定相:烷基、苯基等非极性有机分子。如最常用的ODS柱或C18柱就 是最典型的代表,其极性很小。 适于分离非机性、弱极性离子型样品, 是当今液相色谱的最主要分离模式。正相HPLC(normal phase HPLC): 是由极性固定相和非极性(或弱极性)流动相所组成的HPLC体系。其代表性的固定相是改性硅胶、氰基柱等,代表性的流动相是正己烷。吸附色谱也属正相HPLC。 反相HPLC(reversed phase HPLC): 由非极性固定相和极性流动相所组成的液相色谱体系,与正相HPLC体系正好相反。其代表性的固定相是十八烷基键合硅胶(ODS柱,Octa Decyltrichloro Silane),代表性的流动相是甲醇和乙腈。四、体积排阻色谱(SEC,size exclusion chromatograghy)(又称凝胶色谱和分子筛色谱) 原理: 以多孔凝胶(如葡萄糖,琼脂糖,硅胶,聚丙烯酰胺等)作固定相,依据样品分子量大小达到分离目 的。大分子不进入凝胶孔洞,沿多孔凝胶胶粒间隙流出,先被洗脱;小分子进入大部分凝胶孔洞, 在柱中被强滞留,后被洗脱。根据样品性质分类:凝胶过滤(GFC)—用于分析水溶性样品,如多肽、蛋白、生物酶、寡聚核苷酸、多聚核 苷酸、多糖。 凝胶渗透(GPC)—用于分析脂溶性样品,如测定高聚物的分子量。 SEC主要依据分子量大小进行分离,因此与样品、流动相间的相互作用无关。因此不采用改变流动相的组成来改善分离度。 五、离子交换色谱(ion exchange chromatography, IEC)分离原理:使用表面有离子交换基团的离子交换剂作为固定相。带负电荷的交换基团(如磺酸基和羧酸基)可以用于阳离子的分离;带正电荷的交换基团(如季胺盐)可以用于阴离子的分离。不同离子与交换基的作用力大小不同,在树脂中的保留时间长短不同,从而被相互分离。
有毒气体的分析方法常见的有那四种(1) HCN(2) CO(3)硫化氢(4)氨气的
改善酸峰形的四种途径 色谱工作者时常对碱性化合物拖尾(峰形差)的现象比较熟悉。这种现象通常是由于碱性化合物中的碱性氮原子与固定相中酸性硅醇基的相互作用而至。分析者通常使用加入三乙胺的办法来减少拖尾现象的发生。酸性物质峰形差的现象不多见,但同样给分析结果造成不良影响。下文以布洛芬(对异丁基苯异丙酸)的分析为例,介绍几种改善峰形的方法。 1. 增加流动相中盐的浓度。 分析布洛芬的一种方法是使用60∶40(v/v)的乙腈-5mM磷酸二氢钠。图1(a)中是使用此流 动相所得色谱图,拖尾因子高达(Tf)3.9。高盐浓度(如,25-50mm)磷酸盐可改善酸性物 质的峰形,原因是高盐可抑制溶质与硅胶的离子化以及二者之间的第二相互作用。本例中,分析者应注意分析方法本身及所分析溶质。布洛芬的酸性官能团-羧基(见图1a)的 pka4.4。5mM磷酸二氢钠的pH也是4.4,此pH在磷酸盐的缓冲范围之外(1.1-3.1)。因此, pH4.4时,布洛芬的羧基刚好达到解离平衡(离子化状态与非离子化状态共存)。羧基 (特别是离子化状态羧基)可发生离子交换作用或与硅胶表面的质子竞争,导致拖尾或增加保留。为降低这些相互作用,分析者必须使流动相的pH值远离样品的pka值。 2. 降低流动相的pH值。 将流动相的pH值降低到3.0,峰形得以改善(见图1b)。pH3.0拖尾因子高达(Tf)3.9时,在流动相缓冲范围内,但缓冲能力较低。使盐浓度增加到大于20mM可改善色谱重现性,也能改善峰形。此时布洛芬的羧基不同于pH4.4时的离子化与非离子化混合状态,而 处于单一质子化状态。质子化状态时羧基与硅胶表面质子化的硅醇基相互作用的可能性就小得多。因此,低pH值与盐条件下,布洛芬的拖尾因子降至1.8(图1b)。此结果表明,第二种相互作用已被降低,但尚未消除。另外一种改善峰形的办法及使用添加剂。 3. 加入竞争有机酸。 与加入三乙胺抑制碱性化合物拖尾原理相似,有机酸可与酸性样品组分竞争硅胶表面的活性位置。在流动相中加入1%乙酸对峰形的改善可见图1c,其中得到了对称的布洛芬峰,拖尾因子为1.0。流动相也可再作改变以期获得所希望的结果。 4. 使用另一种有机酸- 0.1%三氟乙酸。 较高浓度的乙酸会产生较高的基线噪音(图1c)。使用0.1%三氟乙酸(约13mM) 取代乙 酸及磷酸盐,使流动相更为简单,仍可获得非常对称的布洛芬峰(图1d)。除此之外,这一流动相还改善了UV透过性,满足了[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]的挥发性需要。

一直以来,实验室的仪器基本是进口的,所用的色谱柱基本也是进口的。日前,拿到了根月旭的极限柱,据说性能不错,是据月旭的销售所说,呵呵。本着新品进实验室必先验证的原则,先验证一番性能,正好,手上正在做黄曲霉毒素的样品,换上后,发现柱子性能还不错。 色谱条件如下: 仪器:WATERS ACCQUITY UPLC 流动相:A:H2O;B:乙腈-甲醇(1:1),梯度洗脱 进样量:10 μL; 柱温:40℃; 黄曲霉毒素四种混标:B1、G1:1ng/ml; B2、G2:0.25 ng/ml; 色谱柱:1、WATERS BEH (2.1*100mm,1.7洀嬀昀漀渀琀=宋体]);2、[font='Times New Roman'][size=12.0pt]WELCH ULTIMATE XB-C18 2.1*100mm,1.8洀。[/size][font='Times New Roman'][size=12.0pt] 原先,仪器上正在使用的是柱1,waters的BEH柱子。在5分钟内,四种黄曲霉毒素全部出峰,能达到完全基线分离。 [/size][font='Times New Roman'][size=12.0pt] [img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471016_1618106_3.png[/img][/size][font='Times New Roman'][size=12.0pt] 换上柱2,极限柱,在相同的梯度条件下,四种黄曲霉毒素的分离情况与柱1基本相同,保留时间推后了大概1分钟的样子,在6分钟内也全部出峰。[/size][font='Times New Roman'][size=12.0pt][img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471017_1618106_3.png[/img][/size][font='Times New Roman'][size=12.0pt] 把两张图叠加,看的更明显。峰高、峰型几乎完全一样。保留时间有所差异。[/size][font='Times New Roman'][size=12.0pt][img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471018_1618106_3.png[/img] [/size][font='Times New Roman'][size=12.0pt] 把四个峰的地方放大,可以看的更清楚。[font='Times New Roman'][size=16px]峰高、峰型几乎完全一样。[/size][/size][font='Times New Roman'][size=12.0pt][img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471019_1618106_3.png[/img] [/size][font='Times New Roman'][size=12.0pt] 分别进一针基质样品(加标),柱1:[/size][font='Times New Roman'][size=12.0pt][img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471020_1618106_3.png[/img] [/size][font='Times New Roman'][size=12.0pt] 柱2:[/size][font='Times New Roman'][size=12.0pt][img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471021_1618106_3.png[/img] [/size][font='Times New Roman'][size=12.0pt] 两张谱图叠加[img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471022_1618106_3.png[/img] [/size][font='Times New Roman'][size=12.0pt] 局部放大后[img]http://ng1.17img.cn/bbsfiles/images/2013/10/201310151419_471023_1618106_3.png[/img] [/size][font='Times New Roman'][size=12.0pt] 两根柱子,内径、长度一样,填料颗粒大小稍有差异,waters的更小一些。[/size][font='Times New Roman'][size=12.0pt] 从分离情况来看,如果不看保留时间,极限柱的分离能力不亚于waters的BEH柱。据说,极限柱的填料完全是国产的,很欣慰,国产的色谱柱也能有如此出色的表现。[/size][size=12.0pt][/size]
各位大神在做甲硫醇等四种气体的时候,那个气体的浓缩使用什么做的呢,方法里用到的样品浓缩管,大家知道哪里有卖的吗?
有谁用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]研究过二甲戊灵、双甲脒、百菌清、甲霜灵这四种农药的残留阿?有的话,用的什么前处理,效果如何?

SPE—HPLC法分离检测水体中四种内分泌干扰物 龚诚,康迪,王猛猛,刁悦,史武男,张郑乐,陈建秋,沈卫阳(中国药科大学分析化学教研室江苏南京210009,药物质量与安全预警教育部重点实验室江苏南京210009)摘要:采用ProElut C18固相萃取柱及HPLC-UVD系统研究了水样中4种代表性的内分泌干扰物(EDCs)的分离检测方法。在水样pH=4.0,用8 mL乙腈为洗脱剂的实验条件下,莠去津、氟虫腈、五氯苯酚和邻苯二甲酸--q一酯的回收率分别为92.6%,92.9%,81.5%,85.9%,线性范围都在50—500岭/L之间,相关系数r均犬于O.9995,检测限分别为1.03×103、1.43 X 10-1、8.12 X 10之、3.46×l o-1 pg·L.1。该方法应用于中国药科大学校尉自来水、池塘水和河水等3处水体的检测,分别检出邻苯二甲酸二丁酯1.8l、2.96、4.05“g·L-1,其余3种物质均未检出,证实该区域增塑剂污染的普遍性和代表性。关键词:固相萃取环境内分泌干扰物高效液相色谱http://ng1.17img.cn/bbsfiles/images/2012/07/201207241334_379378_2355529_3.jpg
工作站打印分析结果 一色谱法也叫层析法,它是一种高效能的物理分离技术,将它用于分析化学并配合适当的检测手段,就成为色谱分析法。 色谱法的最早应用是用于分离植物色素,其方法是这样的:在一玻璃管中放入碳酸钙,将含有植物色素(植物叶的提取液)的石油醚倒入管中。此时,玻璃管的上端立即出现几种颜色的混合谱带。然后用纯石油醚冲洗,随着石油醚的加入,谱带不断地向下移动,并逐渐分开成几个不同颜色的谱带,继续冲洗就可分别接得各种颜色的色素,并可分别进行鉴定。色谱法也由此而得名。1、色谱分离基本原理: 在色谱法中存在两相,一相是固定不动的,我们把它叫做固定相;另一相则不断流过固定相,我们把它叫做流动相。 色谱法的分离原理就是利用待分离的各种物质在两相中的分配系数、吸附能力等亲和能力的不同来进行分离的。 使用外力使含有样品的流动相(气体、液体)通过一固定于柱中或平板上、与流动相互不相溶的固定相表面。当流动相中携带的混合物流经固定相时,混合物中的各组分与固定相发生相互作用。 由于混合物中各组分在性质和结构上的差异,与固定相之间产生的作用力的大小、强弱不同,随着流动相的移动,混合物在两相间经过反复多次的分配平衡,使得各组分被固定相保留的时间不同,从而按一定次序由固定相中先后流出。与适当的柱后检测方法结合,实现混合物中各组分的分离与检测。 气相色谱仪的特点 高灵敏度:可检出10-10克的物质,可作超纯气体、高分子单体的痕迹量杂质分析和空气中微量毒物的分析。 高选择性:可有效地分离性质极为相近的各种同分异构体和各种同位素。 高效能:可把组分复杂的样品分离成单组分。 速度快:一般分析、只需几分钟即可完成,有利于指导和控制生产。 应用范围广:即可分析低含量的气、液体,亦可分析高含量的气、液体,可不受组分含量的限制。 所需试样量少:一般气体样用几毫升,液体样用几微升或几十微升。设备和操作比较简单仪器价格便宜。 气相色谱简单分析装置流程 气相色谱法简单分析装置流程基本由四个部份组成: 1、气源部分,2、进样装置,3、色谱柱,4、鉴定器和记录器.色谱分类方法: 色谱分析法有很多种类,从不同的角度出发可以有不同的分类方法。 ㈠按固定相聚集态分类: 1、气固色谱:固定相是固体吸附剂, 2、气液色谱:固定相是涂在担体表面的液体。 ㈡按过程物理化学原理分类: 1、吸附色谱:利用固体吸附表面对不同组分物理吸附性能的差异达到分离的色谱。 2、分配色谱:利用不同的组分在两相中有不同的分配系数以达到分离的色谱。 3、其它:利用离子交换原理的离子交换色谱:利用胶体的电动效应建立的电色谱;利用温度 变化发展而来的热色谱等等。 ㈢按固定相类型分类: 1、柱色谱:固定相装于色谱柱内,填充柱、空心柱、毛细管柱均属此类。 2、纸色谱:以滤纸为载体, 3、薄膜色谱:固定相为粉末压成的薄漠。 ㈣按动力学过程原理分类:可分为冲洗法,取代法及迎头法三 气相色谱法的常见术语及概念解释 1、相、固定相和流动相:一个体系中的某一均匀部分称为相;在色谱分离过程中,固定不动的一相称为固定相;通过或沿着固定相移动的流体称为流动相。 2、色谱峰:物质通过色谱柱进到鉴定器后,记录器上出现的一个个曲线称为色谱峰。 3、基线:在色谱操作条件下,没有被测组分通过鉴定器时,记录器所记录的检测器噪声随时间变化图线称为基线。 4、峰高与半峰宽:由色谱峰的浓度极大点向时间座标引垂线与基线相交点间的高度称为峰高,一般以h表示。色谱峰高一半处的宽为半峰宽,一般以 x1/2表示。 5、峰面积:流出曲线(色谱峰)与基线构成之面积称峰面积,用A表示。 6、死时间、保留时间及校正保留时间:从进样到惰性气体峰出现极大值的时







 我要推广仪器
我要推广仪器
 下载APP
下载APP