[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102921]多菌灵原药中2个酚嗪类杂质含量的高效液相色谱法测定[/url]
标准品是异黄酮的4种组分,用的流动相是甲醇:水:醋酸=45:54:1,第一和第二种组分的峰型很好,但是第三和第四种组分的峰总是与杂质峰分不开。我也调整了流动相的比例,进样量也调整过了,但是问题依然存在。(我用的液相色谱仪比较老,只能进行等梯度洗脱)。我是新手,对液相色谱了解的也不是很多,现在很着急啊,连标准曲线都做不好。更别说将来要测量样品中的含量了。
对于杂质的含量如何定量?对于固体的杂质含量比较容易定量,但是如果是液体中的杂质定量的话相对来说就比较难了。对于液体来说按面积归一法算出,与稀释倍数有什么关系呢?如何确定呢?
有没有分析3-氨基丙腈的液相色谱分析方法我想用液相色谱分析有机盐中的3-氨基丙腈的含量,但由于3-氨基丙腈在紫外和示差液相中响应值不是很大,满足不了微量杂质的分析。大家有没有好的建议。比如有没有合适的检测器、或则有合适的衍生法。(由于样品中含有氨基丙酸,所以氨基的衍生手段好像无法使用)
:高效液相色谱_串联质谱法测定酸奶中纳他霉素的含量期刊:中国卫生检验杂志作者:李媛媛; 王伟; 关树文; 李刚; 董巧红;链接:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=ZWJZ201004027
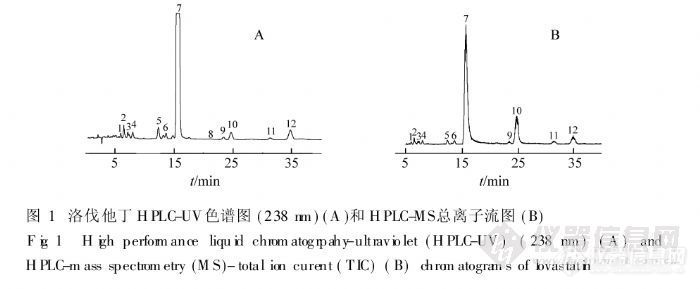
【作者】 吴永江; 朱炜; 邵青; 程翼宇;【Author】 Wu Yongjiang,Zhu Wei,Shao Qin,Cheng Yiyu~*(College of Pharmaceutical Sciences,Zhejiang University,Hangzhou 310031)【机构】 浙江大学药学院; 浙江大学药学院 杭州310031; 杭州310031;【摘要】 利用高效液相色谱-二极管阵列检测器-质谱联用方法对洛伐他丁及其杂质成分进行分离分析和结构鉴定。实验采用D iamonsil C18(5μm,4.6 mm×250 mm)为分离柱,乙腈-水(含0.1%乙酸)(65∶35)为流动相,分离并检测了洛伐他丁及其杂质;通过与DAD检测器和离子阱质谱联用,获得了它们的紫外光谱和质谱数据;紫外光谱表明除氢化洛伐他丁外其余杂质与洛伐他丁基本结构相同,利用MS和MS2数据确定了杂质的分子量和侧链结构,由此鉴定了其中10个杂质的结构。实验结果表明,高效液相色谱-二极管阵列检测器-质谱联用技术可以快速鉴定洛伐他丁中的杂质化学成分。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301723_380642_2379123_3.jpg
用液相色谱检测控制杂质限量,要求是杂质含量超过0.1%的不能超过三个。但是设定不同的积分参数(峰高,峰面积,峰宽,斜率等),得到的杂质峰个数可能就不一样,请问大家在遇到这种情况的时候是怎么做的?对此有没有标准文件可以参考的?还是全凭经验来?个人有个想法就是根据仪器检测限,即3倍信噪比来确定峰高这一参数,不知道合理不合理?请个人大神指教!
氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
我做了一个氨氯吡啶酸(毒莠定)的制剂,用液相色谱测定含量,结果测出的含量总是偏低,我用我现配的样品去测还是还是这个问题。我们实验室的两个人配样品,两个人测,结果都这样。峰形出的很好,和杂质是基线分离。线性相关性有0.9985,测出回收率有在99%左右。我测过原药的含量,原药没问题,样品分解的可能性不大。有哪位高手遇到过这种问题,请指教一下。谢谢了!!
请大家帮忙下载一下这篇文献"高效液相色谱法测定蓝萼甲素的含量"本人急需. 由于这篇文献出自1990年解放军医学杂志,太老了本图书馆检索不到.请求大家的帮助.
如题。做液相色谱质谱杂质峰丰度很高,达到2*E5,怎么冲都冲不掉,做的是蛋白酶解实验。各位有办法吗?又遇到类似情况吗?
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]含量测定中测的值为98%,实际含量为96%,一个称量0.1g,稀释100倍,一个称量0.01g稀释10倍,结果前者测得含量98,后者测得含量97,这个是什么原因

今天接到客户送来的样品,让我们检测一下样品中是否含有杀铃脲,如果含有含量是多少?老规矩先要了解一下杀铃脲,它是一种接触性和胃毒性杀虫剂,属于苯甲酰脲类的昆虫生长调节剂,阻止了昆虫体内合成,使昆虫幼虫虫体畸形而死亡。主要用来防治玉米、棉花、森林、水果和大豆上的鞘翅目、双翅目、鳞翅目害虫,对此类害虫的天敌却无害。此种杀虫药属于低毒杀虫剂,并且效率高,对大多数动物和人无毒害作用,从而被广泛使用。 本次试验将试样用二甲基甲酰胺溶解,通过液相色谱仪,以C18柱为固定相,甲醇+水为流动相,将有效成分与杂质分开,在波长为254nm下,用紫外检测器,数据处理系统,测定流出峰面积,用外标法,定量有效成分的质量分数。一、[b]实验试剂[/b]杀铃脲标准品:已知质量分数为97%;N,N—二甲基甲酰胺:分析纯;水:新蒸二次蒸馏水;甲醇:色谱纯。二、[b]实验仪器[/b]高效液相色谱仪:具有 254nm 波长紫外检测器[img=,375,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221113548365_8139_3763765_3.jpg!w690x791.jpg[/img]色谱柱:C18不锈钢柱[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221115252987_2106_3763765_3.jpg!w690x1226.jpg[/img]微量注射器:50μl。[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221116089617_8904_3763765_3.jpg!w690x1226.jpg[/img]流动相过滤器[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221117154197_3452_3763765_3.jpg!w690x1226.jpg[/img]电子天平[img=,297,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221118120157_7469_3763765_3.jpg!w690x998.jpg[/img]超声波清洗器[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221118523747_2538_3763765_3.jpg!w690x1226.jpg[/img]三、[b]色谱条件[/b]流动相:甲醇:水 ﹦80:20流量:1.0ml/min检测波长:254nm进样体积:20μl四、[b]样品前处理[/b]流动相:将甲醇和水按8:2比例混合,经过流动相过滤器过滤后,转移至棕色瓶中,超声后备用。标准品溶液:称取杀铃脲标准品5mg(精确至 0.0002g)于50ml容量瓶中,加入约22ml二甲基甲酰胺溶解,在超声中脱气10min,定容后摇匀。样品溶液:称取样品100mg(精确至 0.0002g)于5ml 容量瓶中,加入约2ml 二甲基甲酰胺溶解,在超声波中脱气 10min,定容后摇匀,用0.5μm孔径过滤器过滤,待测五、[b]测定[/b] 打开机器,设定好上述参数,排气完毕后,待基线走平稳后,使用进样针连续注入三针标准品后,标准品峰面积变化相差小于0.5%,随后按照标准品,样品,样品,标准品的顺序依次进样检测。六、结果及讨论 将测得的两针样品溶液以及样品前后两针标准品溶液的峰面积,分别进行平均,按照下式进行计算: [img=,130,66]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221119280279_4828_3763765_3.png!w130x66.jpg[/img]C样:样品浓度C标:标准品浓度A样:样品平均峰面积A标:标准品平均峰面积[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221123338764_2616_3763765_3.jpg!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221124134784_89_3763765_3.jpg!w690x387.jpg[/img] 根据谱图判定样品中含有杀铃脲,计算出样品中杀铃脲的含量为:4mg/100mL。
液相色谱,有结晶水的原料药怎样计算含量我们液相色谱测样品的含量,样品中含两个结晶水,但所用对照品是无水的,计算样品含量时要扣除那两分子的水分吗?
请问怎么用液相色谱测定样品中的奈含量色谱仪不限 样品不限最好能附操作步骤谢谢大家!!
如题,请知道的朋友告诉我,先谢谢了! 我即将参与做的一个3.1类新药项目,其杂质定量研究正需要考虑这个响应因子的问题,以前做的都是补充申请,还没有考虑响应因子不一致的情况,现在考虑了一个方案,那就是液相色谱检测,液相色谱和紫外同步进行,如果我们这个新药项目杂质可得,并且有对照品,那么就可以绘制杂质和对照品的标准曲线,杂质标准曲线的斜率除以对照品标准曲线的斜率的比值就是响应因子吧?我想参照指导原则确定到底什么范围时可以用主成分的自身对照法计算含量,什么范围时,宜用杂质对照品法计算含量,也可用加校正因子的主成分自身对照法?您有好的建议吗?期待您的帮助。
液相色谱法测定乳制品中苯甲酸含量摘要: 使用液相色谱仪检测乳制品中苯甲酸防腐剂含量。关键词:乳制品;液相色谱;苯甲酸;防腐剂参考文献:GB 21703-2010苯甲酸是食品工业中常见的一种防腐保鲜剂,但乳制品中是禁止添加的。一般情况下,苯甲酸被认为是安全的,但对包括婴幼儿在内的一些敏感人群而言,长期摄入苯甲酸也可能带来哮喘、荨麻疹、代谢性酸中毒、抽搐等不良反应。实验部分1试验材料、仪器及试剂1 样品:新疆奶牛基地生鲜乳2 仪器:液相色谱仪-Waters 2695配紫外检测器;天平(0.01g)(奥豪斯(上海)有限公司);3试剂:氢氧化钠、浓硫酸、亚铁氰化钾、乙酸锌(分析纯,天津盛奥化学试剂厂);甲醇(色谱纯,Therm Fisher)2 标准品配制:苯甲酸标准贮备液:500mg/L;标准工作液:10mg/L。3 样品前处理 称取乳制品液体试样20g±0.01置于100mL的容量瓶中,加入25mLNaOH(0.1mol/L),混匀振荡15min,用H2SO4(0.5mol/L)调节pH≈8,再加入2mL亚铁氰化钾(0.25mol/L)和2mL乙酸锌(1mol/L),混匀振摇,静置后用甲醇-水(v/v=1/1)定容至100mL,上清液过0.45μm滤膜过滤后装进样瓶待测。4 仪器条件色谱柱:C18柱(Waters);流动相:甲醇-磷酸盐缓冲溶液=1:9;流速:10mL/min;检测波长:227nm;进样量:10uL。5 结果与分析 http://ng1.17img.cn/bbsfiles/images/2014/07/201407071107_505678_1634341_3.bmp[
各位老师,我现在在做硝苯地平的有关物质,有一个硝苯地平杂质1的含量忽大忽小,后来我们发现不同的色谱柱,检测出来的含量不同,用了安捷伦、迪马家的C18柱,杂质1含量约0.3%,但是用Waters家的色谱柱,杂质1就是未检出。所以,我想问一下,C18色谱柱中,会不会存在微量物质,促使硝苯地平降解为杂质1。硝苯地平在紫外光破坏、酸破坏、热破坏条件下产生杂质1。另外,单独进杂质1对照品溶液峰面积无变化,每个色谱柱都很稳定。
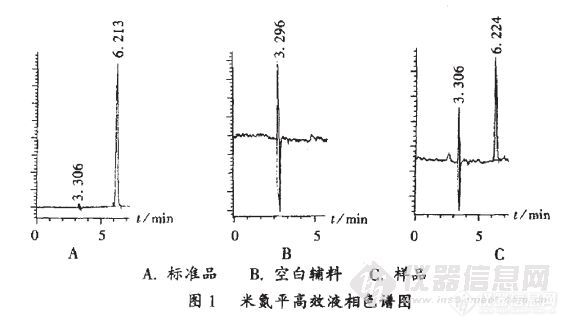
作者:温预关, 王玲芝, 刘学军, 陆欣乔(广东省广州市脑科医院临床药理实验室, 广东 广州 )摘要:目的:建立测定米氮平片中米氮平含量的反相高效液相色谱法。方法:以美国Dikma公司DiamonsilTMC18反相柱(250mm×4.6mm,5μm)为色谱柱,流动相为甲醇-超纯水(90∶10,V/V),流速为0.8mL/min,检测波长为294nm,柱温为30℃,进样量为5μL。考察了流动相不同配比对米氮平色谱行为的影响。结果:米氮平与片剂辅料及其杂质可完全分离;分析方法的定量测定下限为0.2μg/mL,线性范围:1.0~100.0μg/mL,回归方程为C=1.85×10-1F+9.65×10-1,r=0.999(n=9),平均回收率为95.15%,RSD=0.76%。结论:方法灵敏、准确、简单、快速,可用于米氮平片的含量测定。http://ng1.17img.cn/bbsfiles/images/2012/07/201207161639_377909_2379123_3.jpg
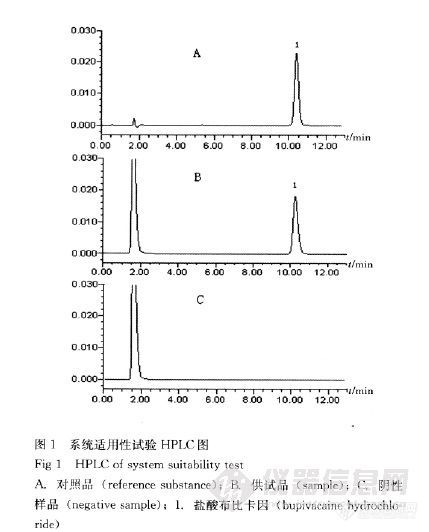
作者:兰文; 杨汉初; 黄莉;(湖南省药品检验所;)摘要:目的建立反相高效液相色谱法测定复方明矾布比卡因注射液的有关物质及盐酸布比卡因的含量。方法采用Diamonsil C18柱,以乙腈-磷酸盐缓冲液(取磷酸二氢钾1.94g,磷酸氢二钾2.48g,加水溶解并稀释至1 000mL,调节pH 6.8)(65∶35)为流动相;流速为1.0mL.min-1;有关物质检测波长为215nm,含量测定检测波长为263nm;柱温:30℃。结果在该色谱条件下,杂质峰与主峰均能有效分离,盐酸布比卡因在44.31~177.26μg.mL-1与峰面积线性关系良好(r2=0.999 7);平均回收率为99.7%(n=9),RSD=0.4%。结论本法简便、快速、准确,专属性好,可用于复方明矾布比卡因注射液的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061043_381724_1606903_3.jpg
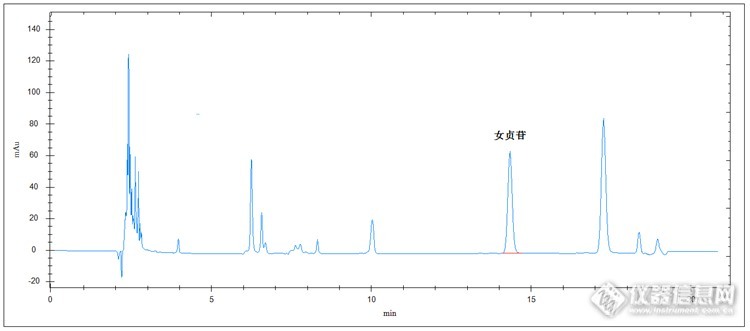
高效液相色谱法检测女贞子药材中女贞苷含量 药材女贞子为木樨科植物,具有滋肝肾、明目黑发,也可治疗眩晕耳鸣、腰膝酸软等症状。其中女贞苷是女贞子中重要成分之一。相应饮片和药材都要求检测女贞苷含量。 以下的方法采用的是高效液相色谱法,检测的是女贞子药材中女贞苷含量。实验部分原理 取适量样品粉碎、溶解、超声提取、定容、滤过,由进样系统进样,色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量计算。仪器及试剂 仪器:高效液相色谱仪(紫外检测器),超声波清洗仪,粉碎设备,三号筛,电子天平,具塞锥形瓶,溶解过滤器,针筒式过滤器 试剂:甲醇(色谱纯),超纯水,稀乙醇样品制备 对照品溶液的制备:精密称取女贞苷对照品12.5mg于50ml容量瓶中,加甲醇制成0.25mg/ml女贞苷溶液,备用。 供试品溶液的制备:取女贞子药材样品适量,充分粉碎后过三号筛,准确称取过筛后的粉末0.5g,置于具塞锥形瓶中,精密加入稀乙醇50ml,称定重量,超声波超声30min,待样品完全溶解后,放冷,再称定重量,用稀乙醇补充减少的重量,摇匀,微膜滤过,待测。色谱条件检测器:紫外检测器色谱柱:C18,4.6 X 250mm,5um波长:224nm流动相:甲醇:水=40:60(V:V)流速:1.0ml/min柱温:30℃进样量:5ul对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_518997_2498430_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_518998_2498430_3.png 看看这效果,很羡慕吧,峰形、分离度都很完美吧。 该方法检测女贞子药材中女贞苷含量操作简单、方便,实验快速,结果准确、可靠,是一种坚持女贞子的好方法。 注意的是样品前处理时如果取样量较大或样品粉碎的不够充分,样品提取时可能不能提取完全。那样的话一是样品量要适中,不要太多;二是样品粉碎的要尽量充分;三是超声波提取时间要适当长一些,或提取时温度湿度高一点,或分成几次或几批提取等。 另外这个样品杂质和目标物干扰物都较少,检测时可以适当考虑下缩短检测时间的问题,比如适当增加流动相中甲醇含量,适当升高色谱柱温度控制等。下面我们就看看流动相甲醇:水=50:50,柱温40℃的效果吧。对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_518999_2498430_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_519000_2498430_3.png 效果很好吧,这种方法我们在检测时是不是可以考虑下。哈哈,经验之谈,祝大家工作顺利,天天开心!
求用高速液相色谱法测植物激素含量如题本人想用高效液相色谱法测定花芽中赤霉素的含量想知道实验之前要做怎样的准备工作求助大家谢谢啦
[b][color=#444444]请问各位大侠,谁有用液相色谱测浓缩果汁中乳酸含量的检测方法?急需!谢谢![/color][/b]
请教:液相色谱测定原料药杂质如果结果分别在5%、2%、1%、0.5%、0.1%以内平行结果的相对误差分别应在多少以内?如果是不同仪器、不同色谱柱、同样方法相对误差又该是多少呢?
高效液相色谱分析血液中儿茶酚胺含量,回收率不高各位老师,我想要用用高效液相色谱分析血液中儿茶酚胺含量,用的是电化学检测器,柱子是C18柱,用氧化铝吸附解析,找了几篇论文但是回收率不高,不知道是柱子没选对,还是前处理做的不好,或者是流动相不好,各位老师有什么意见可以知道一下吗?
应用液相色谱法测定生物制品中甲醛的含量 摘要 液相色谱测定甲醛与2, 4-二硝基苯肼的反应条件, 使反应在菌疫苗类生物制品自身的pH 范围内即弱酸性条件下, 无需酸碱度调节, 反应产物甲醛衍生物不需要有机溶剂萃取, 避免损失, 可直接用于液相色谱分析。在检出限及精确度方面更是优越于分光光度法。结果显示, 该研究方法的色谱条件能准确定量、灵敏、准确、重复性好, 优于药典的分光光度法, 可用于实际检测分析。
有没有做过维生素A杂质的液相色谱分析?维生素A及其杂质维生素A环氧化物、维生素A醛、维生素A酸没有杂质对照品,以上物质在甲醇-水(90:10)流动相,C18*250mm柱能不能分的开?
[align=center][b]浅谈药典中高效液相色谱法测定中药材含量方法学验证的方法[/b][/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]食品事业部:任阳阳[/align]药典中关于方法学验证有相应的指导原则,指导原则上写的内容较宽泛和笼统,但对于大多数检测人员来说根本不知道如何下手,本文是根据自己的理解和经验来阐述如何做好药典中高效液相色谱的方法学验证。首先要了解做好药典中高效液相色谱的方法学验证,要好好的理解两部分的内容:第一,要熟悉0512高效液相色谱法这部分的内容;第二是理解9101药品质量标准分析方法验证指导原则,结合这两部分内容,做好方法学验证。1. 确定本实验室要使用的仪器和配件能否满足本次验证的色谱提交,包括色谱柱、检测器和流动相。2. 0512方法中验证系统适用性实验首先要了解色谱的适用性实验包括5个参数:理论塔板数、分离度、灵敏度、拖尾因子和重复性。2.1理论塔板数和分离度在已调试好的色谱条件下,分析目标物质,根据分析结果计算此方法的理论塔板数和分离度。一般待测物质色谱峰与相邻色谱峰之间的分离度不大于1.5。2.2 灵敏度通过一系列浓度的对照品确定信噪比,其灵敏度要求定量测定时信噪比≥10;若为定性鉴别其信噪比要求≥3。2.3拖尾因子T拖尾因子是评价色谱峰的对称性结果,以T表示,对对照品进行进样,分析色谱结果,以峰高作定量参数时,T值应在0.95~1.05之间。2.4 重复性 一般采用外标法,取对照品溶液连续进样5次,计算5次进样的峰面积的相关标准偏差,要求≤2.0%以上5点是0512方法内容中需要验证的5点内容,已对方法进行了简单的验证,但这些不够,还得结果9101的内容进行详细的进一步的验证。3. 9101验证的指标主要有准确度、精密度、专属性、检出限、定量限、线性和耐用性,其中精密度可以参考0512中已验证的重复性的检测结果;检出限和定量限也可参考0512中验证的结果。3.1准确度准确度是指该方法测定结果与真实值或参考值的接近程度,常用回收率进行验证。一般选择高、中、低三个浓度进行测定,也就是说,对照品加入量与待测供试品含量的比值为1.5:1,1:1和0.5:1进行加标实验,分析加标结果,计算回收率和回收率的相对标准偏差,结果必须满足药典要求。3.2 专属性向试样中加入杂质或辅料,与杂质和辅料的结果进行比较,确定测定结果是否受干扰。3.3 线性根据待测成分的含量大小,选择至少5份合适的浓度范围进行分析,以测得的响应信号(峰高或峰面积)对被测物的浓度作图,求出线线方程和相关系数。3.4耐用性一般经过变动流动相的比例、流动相的pH、不同类型的色谱柱。柱温以及流速来进行验证,验证测定条件发生微小变化时,测定结果不受影响的程度,从而确保方法的可靠性。综上所述,以上是高效液相色谱法测定中药材含量的方法学验证的所有指标,通过以上指标的完善和验证,才能做出一套完成的方法学验证。
利用高效液相色谱仪检测鱼肉中ATP等含量,由于样品处理时间较长,ATP容易分解,有没有什么比较快的样品前处理的方法?
谁有 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定氯气氢气中杂质含量 的分析方法的相关资料,我从网上搜了一下,都是万方收费的 大家有没有免费的资料 谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP