超短链全氟烷基化合物“三氟乙酸”分析利器——超临界流体色谱质谱联用技术
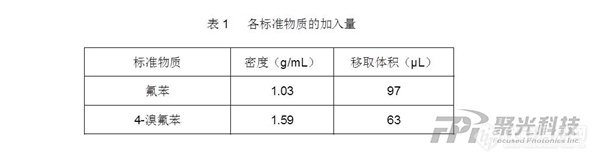
近年来,以三氟乙酸(TFA)为代表的超短链全氟烷基化合物(超短链PFAS)大量赋存于城市河水中这一问题已对城市生态及饮用水生产带来了巨大挑战,监测和精确定量饮用水源中的超短链PFAS已经迫在眉睫。针对高极性的超短链PFAS,高效环保的超临界流体色谱质谱联用技术可以提供良好保留和高灵敏度检测结果。背景介绍PFAS是一类广泛用于消费品和工业生产的含氟有机化合物。全氟辛烷磺酸(PFOS)和全氟辛酸(PFOA)是两种含八个碳的全氟烷基酸类化合物(PFAA),因具有较高的环境持久性和毒性,已在全球范围内逐步淘汰。然而,取而代之的是一些超短链(C1&minus C3)(图1)和短链(C4&minus C7)PFAA,其在环境、血液及尿液样本中正在被广泛检出【1,2】,引发了人们对健康影响的担忧。图1 超短链(C1&minus C3)全氟烷基化合物特别是含量较高的三氟乙酸被认为含有损坏生育能力和儿童发育毒性,正在全球范围内引起广泛关注。据欧洲新闻网报道,欧洲农药行动网络(PAN Europe)及其成员于5月27日联合发布了一项研究报告,对来自10个欧盟国家的23个地表水样本和6个地下水样本的联合调查发现,所有检测的水样中均检测到PFAS,其中23个样本(79%)的TFA浓度超过了欧盟饮用水指令中“PFAS总量”的拟议限值;而在检测到的总PFAS中,TFA占总量的98%以上【3】。TFA是含有两个碳的全氟羧酸,属于超短链(C1&minus C3)全氟烷基化合物。其在环境中普遍存在,主要来源包括PFAS农药、氢氟碳化物制冷剂、污水处理和工业污染(图2)。尽管目前对TFA的生物毒性效应研究有限,考虑到其持久性和全球传播特性,正在引起全球多国的密切关注【4,5】。图2 杀虫剂、杀菌剂和药品中的碳键全氟甲基在环境条件下通过氧化裂解转化为TFA特色应用方案使用高效环保的超临界流体色谱(SFC)分离技术,结合超高灵敏度三重四级杆质谱检测器,岛津中国创新中心开发了包括TFA在内的五种超短链PFAS快速分析方法。与反相液相色谱不同,SFC可以充分保留仅有一到三个碳的超短链PFAS,有效降低基质的干扰(图3)。图3 SFC-MS/MS和LC-MS/MS分析超短链PFAS色谱对比图(1ng/mL标液)使用SFC-MS/MS对纯水配置的系列标准溶液进行分析,可得到良好线性和较低检测限(见表1),进一步,对不同地表水样品进行检测,结果发现,均检测到一定量TFA,使用内标法定量,分别为几百个到几千个ppt,说明TFA在城市水体都存在较为严重的污染(图4、图5)。图4 SFC-MS/MS分析地表水样品1中超短链PFAS图5 SFC-MS/MS分析地表水样品2中超短链PFAS表1 SFC-MS/MS分析水样中超短链PFAS线性和检出限总结采用超临界流体色谱串联三重四极杆质谱仪(SFC-MS/MS)建立超短链(C1&minus C3)全氟烷基化合物的快速分析方法。由于超临界流体色谱独特的分离选择性,使用SFC-MS/MS分析种类繁多的PFAS,可以得到与反相色谱截然不同的溶出顺序和出峰行为。SFC-MS/MS可作为反相液相色谱质谱联用技术一种有力补充,对超短链PFAS进行更准确定量。随着对PFAS及其降解产物(TFA等)认识的不断深入,全球各国需要加强对这些持久性化学品的监管和限制, 旨在减少PFAS污染,保护生态系统和人类健康。超临界流体色谱串联三重四极杆质谱仪(SFC-MS/MS)注解*:超临界流体色谱(SFC):使用超临界流体作为流动相的色谱分离技术。以超临界流体CO2为流动相的SFC分离技术不仅高效而且节能环保,作为一种绿色分离技术在制药、食品和石油领域得到越来越广泛的应用。参考文献1. Guomao Zheng, Stephanie M. Eic, Amina Salamova. Elevated Levels of Ultrashort- and Short-Chain Perfluoroalkyl Acids in US Homes and People. Environ. Sci. Technol. 2023, 57, 42, 15782–15793.2. Isabelle J. N., Daniel H., Hanna L. W., Vassil V., Ulrich B., Karsten N., Marco S., Sarah E. H, Hans P. H. A., and Daniel Z., Ultra-Short-Chain PFASs in the Sources of German Drinking Water: Prevalent, Overlooked, Difficult to Remove, and Unregulated. Environ. Sci. Technol. 2022 56, 10, 6380-6390.3. 欧洲水体中的PFAS污染引发关注:塞纳河等河流中令人惊讶的三氟乙酸浓度.【微信公众号:新污染物监测与分析】4. Cahill, T. M. Increases in Trifluoroacetate Concentrations in Surface Waters over Two Decades. Environmental Science & Technology, 2022, 56,9428-9434.5. Thomas M. Cahill. Assessment of Potential Accumulation of Trifluoroacetate in Terminal Lakes. Environ. Sci. Technol. 2024, 58, 6, 2966–2972.本文内容非商业广告,仅供专业人士参考。






























 我要推广仪器
我要推广仪器
 下载APP
下载APP