推荐厂家
暂无
暂无
 400-860-5168转1867
400-860-5168转1867
 留言咨询
留言咨询
留言咨询
留言咨询
留言咨询
留言咨询
 400-860-5168转1867
留言咨询
400-860-5168转1867
留言咨询
 400-612-9980
留言咨询
400-612-9980
留言咨询
 400-612-9980
留言咨询
400-612-9980
留言咨询





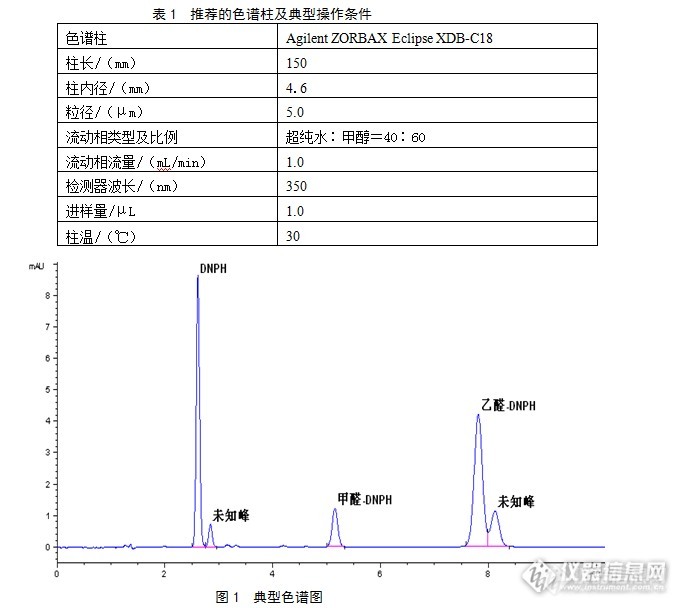
微量甲醛、乙醛液相色谱测定法1 适用范围本标准规定了液相色谱测定水溶液或环氧乙烷样品中的甲醛和乙醛的方法。本标准适用于样品中甲醛、乙醛含量为1~50mg/kg的分析。2 方法概要 液态样品中微量甲醛、乙醛与2、4-二硝基苯肼衍生生成甲醛-2、4-二硝基苯腙(以下称:甲醛-DNPH)和乙醛-2、4-二硝基苯腙(以下称:乙醛-DNPH),在通过固相C18小柱将甲醛、乙醛的衍生物从溶液中萃取出来。随后,使用自动进样器直接将萃取出的甲醛、乙醛衍生物注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],样品在甲醇和水混合溶液携带下通过毛细管色谱柱分离,使样品里的甲醛衍生物、乙醛衍生物及未反应2、4-二硝基苯肼按照不同顺序先后进入紫外检测器检测。最后,由色谱数据工作站采用外标法得到各物质含量。3 试剂和材料3.1 甲醇:液相色谱(HPLC)纯。3.2超纯水:比电阻率大于18.2ΜΩcm的水。3.32,4-二硝基苯肼(DNPH):分析纯。3.4超声波振荡器。3.5玻璃溶剂过滤器。3.6真空泵。3.7量筒:10mL。3.8量筒:25mL。3.9具塞碘量瓶:250mL。3.10C18固相萃取小柱(SPE)。3.11注射器:5mL。3.12注射器:10mL。3.13烧杯:50mL3.14标准溶液:与分析样品浓度相接近甲醛、乙醛水溶液,自配或外购。4 仪器4.1液相色谱仪:安捷伦科技公司1200型,配有自动进样器、真空脱气、四元梯度泵和紫外检测器,或同类产品。4.2色谱数据工作站:安捷伦科技公司Chemstation工作站,或同类产品。4.3色谱柱:见表1推荐色谱柱,或同类产品。推荐色谱柱典型操作条件见表1,典型色谱图见图1。[img=,681,608]http://ng1.17img.cn/bbsfiles/images/2017/09/201709032112_01_2166779_3.png[/img]5 校正仪器第一次使用或者被改变了某一或某些参数设置(如流量、流动想类型、流动相比例、色谱柱等),应对仪器进行校正。使用已知与样品接近含量的标准甲醛、乙醛溶液与DNPH络合后,在液相色谱仪上分析,测量两物质色谱峰峰面积,计算校正因子k[sub]i[/sub]:k[sub]i[/sub]=标准液中i物质浓度/i物质衍生物峰面积。6 试验步骤6.1用5mL注射器将5mL甲醇注入C18固相萃取小柱,冲洗萃取小柱进行预处理,然后再用5mL超纯水冲洗。6.2分别取10g样品(由于环氧乙烷沸点较低,环氧乙烷产品可直接使用量筒量取11.2mL)、10mL2,4-二硝基苯肼(DNPH)试剂和10mL超纯水转移到碘量瓶内,盖上瓶塞,让混合物反应15分钟。如果观察到沉淀,则应使用超纯水按1:10稀释样品,并按上述描述再次进行操作。6.3使用一个10mL量筒量取10mL的反应混合物,用针筒将其转移到一个6.1中预处理好的固相萃取小柱,注射时应将样品缓慢地推进萃取小柱。然后,使用10mL超纯水重复上述步骤,注意需将所有液体推出萃取小柱。6.4用注射器将8mL甲醇推进萃取小柱,以洗脱富集被固相萃取小柱从样品中萃取出的甲醛-DNPH、乙醛-DNPH及未反应的DNPH溶液。洗脱富集液放置于烧杯中。再向烧杯添加少量甲醇,使溶液的最终重量达到10±0.01g。6.5使用符合4.3中推荐色谱柱或同类色谱柱,采用自动进样器将适量6.4中的洗脱富集样品注入4.1规定液相色谱仪,通过色谱工作站测量各物质出峰面积,利用已测校正因子面积归一定量计算样品中各组成含量。6.6用10mL的去离子水替代样品重复步骤6.2至6.5,测定空白。7 计算7.1溶液甲醛、乙醛含量按下式计算:(外标法)X[sub]i[/sub]=A[sub]i[/sub]×k[sub]i[/sub]式中:X[sub]i[/sub]——被测组分i的含量,mg/kg;A[sub]i[/sub]——被测组分i的衍生物峰面积;k[sub]i[/sub]——被测组分i的绝对校正因子;i——甲醛或乙醛。8 精密度8.1 重复性同一操作者重复测定的两次测定结果的差值应不超过平均值的10%(m/m)。9 结果的表示仪器稳定状态下连续两次测定结果的平均值作为出厂产品的测定结果,生产装置过程控制使用单次测定数据作为结果报告,结果的表示修约至1mg/kg。10注意事项10.1 仪器进样前取样,需摇晃采样容器,以保证分析样品的均匀性。10.2 色谱流动相必须使用液相色谱(HPLC)纯试剂。10.3当分析环氧乙烷或含有环氧乙烷样品时,使用前应事先将试剂、容量瓶和量筒冷却,以防止环氧乙烷受热挥发。10.4 对含有环氧乙烷的样品的任何操作,必须在通风柜中完成,并做好个人防护。
液相色谱进样量一般在10微升到20微升之间,这是最常见的进样体积。如果分析对象的灵敏度较低,进样量可以增加到50微升甚至100微升。此外,超高压液相色谱的进样量通常在0.5微升到1微升之间,这是因为超高压液相色谱的柱子与普通液相色谱的柱子不同,进样量少且不好取样。? 进样量的选择需要考虑色谱柱的容量和溶剂效应。进样量一般与色谱柱的容量有关,过多的进样量可能会导致色谱峰的扩散,影响峰面积的准确性。因此,在选择进样量时,需要综合考虑分析物的性质、色谱柱的类型和容量以及实验的具体要求。

液相色谱手动进样阀的使用和保养 液相色谱进样器有手动和自动两种,随着系统自动化的推进,自动进样器已渐趋普及。但是手动进样阀的使用量还是比较大的,尤其是在企业中。我们平时在使用手动进样阀进样时,一般并没有特别留意,但,如果使用方法不当,也会引起色谱图上出现不良峰型、做标准曲线时线性关系差、产生漏液等现象,导致分析出现异常。这里以7725型手动进样器为例,简述避免故障,正确使用的注意事项。7725型手动进样阀[img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909171652_171614_1620630_3.jpg[/img]7725型手动进样阀的结构和原理[img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909171653_171615_1620630_3.jpg[/img]注:以上图片是网上下载的。一、结构 7725型手动进样阀是目前使用最广泛的高压平面进样阀。其结构如上图所示。它由定子、转子密封垫、转子、手柄等组成,在定子上刻有6个号码,在安装时,将5号和6号用附属的管路连接。注意:2号必须与泵相连接,3号必须与柱相连接。在 (load)的状态(手柄向左旋)下,流动相不经过定量环直接进入柱,此时用微量注射器注入样品,样品液流入定量环,过量的样品液从排液管(废液管)流入废液瓶。接着进样(手柄向右旋)处于(Inject)时,由于转子的旋转,定量环与3号排出管路相接,定量环中的样品液被送入柱中。 二、进样操作 进样的方法有环路全量进样和部分进样两种。标准附属的定量环为20µ l或10µ l,如果用约50~100µ l的微量注射器注入全量的话,则环路被清洗,并且环路被样品完全充满。如果用此方法进样,则属于“环路进样”能保证有20µ l样品量注入,这样做重现性较好。这就是全量进样的方法。一般定量分析均采用此方法进样。 部分进样的方法是在以环路容量作为最大的范围下,通过微量注射器注入样品进行进样。采用此方法微量注射器的计量误差将严重影响重现性,但也有其有利性,样品的配制浓度可以较高,尤其适用于定性分析。不论何种方法,使用7125型进样器时,不必取下微量注射器,转动把手即可进样。为了保持针端的密封形状准确,平时请将微量注射器或附带的针插上。 三、进样器的清洗和保养 留在进样阀注射针导入口的残余样品易引起交叉污染(Cross Contamination),在重复进样时会给工作带来影响。在正常情况下不必每次进样后都冲洗进样阀注射针导入口。但是,在进样针头插入或拔出过程中,会有痕量的样品沉积在针头密封区域,这样,就需要在工作结束时或进样间歇时,用附带的聚四氟乙烯针和注射头注入水或甲醇、乙腈清洗进样阀注射针导入口,此时进样阀手柄应向右转处于(Inject)位,注意不要把针头插入。 注意:当进过高浓度样品时,更要及时清洗。四、漏液的产生原因和处理1、长期使用后,因为转子的旋转,其与定子的摩擦,会产生转子密封垫的磨损,产生漏液。此时漏液亦可能向其他流路泄漏,导致色谱图的异常。处理方法:更换7725型的转子密封垫。2、密封垫表面有划痕。在更换进样阀密封圈时,你会发现密封圈上有细微的划痕。漏液就是由这些划痕引起的。这是因为,有些微量注射器的表面不够光滑(肉眼是看不出的),进样时,在密封圈上留下的。建议:使用进口或质量好的微量注射器,以减少进样阀密封圈的受损几率和更换频率。还有,当流动相中存在微小的固体时,或者是盐析出,都会在进样阀密封圈表面留下划痕。因此,流动相要在使用前过滤;使用含盐的缓冲液作流动相时,要注意盐的析出,并及时用水冲洗。五、其它1、耐压性 7725型的耐压为350Kgf/cm2。通常这已经足够了,但如有特殊的使用目的,可以通过增紧定子螺栓来达到500Kgf/cm2的耐压规格。不过为延长使用期,建议在较低耐压规格下使用为宜。2、密封圈材质 一般,7725型的密封材料使用的是被称作BESPEL的聚酰亚胺,因此不可用于pH10以上的碱性流动相。此外,自动进样器SIL-10A、10Avp上用的也是BESPEL。








 我要推广仪器
我要推广仪器
 下载APP
下载APP


