98%,便于与纯化合物的标准进行对照。多组分试样应在测定前尽量预先用分馏、萃取、重结晶、区域熔融或色谱法进行分离提纯。(2) 试样中不应含有游离水。水本身有红外吸收,会严重干扰样品谱,而且还会侵蚀吸收池的盐窗。(3) 试样的浓度和测试厚度应选择适当,以使光谱图中的大多数吸收峰的透射比处于10%~80%范围内。包括控制浓度和压片的厚薄尺寸。2.制样方法(1) 固体样品的制备a.压片法:将1~2mg固体试样与200mg纯KBr研细混合,研磨到粒度小于2μm,在油压机上压成透明薄片,即可用于测定。b.糊状法:研细的固体粉末和石蜡油调成糊状,涂在两盐窗上,进行测试。此法可消除水峰的干扰。液体石蜡本身有红外吸收,此法不能用来研究饱和烷烃的红外吸收。(2) 液体样品的制备a. 液膜法: 对沸点较高的液体,直接滴在两块盐片之间,形成没有气泡的毛细厚度液膜,然后用夹具固定,放入仪器光路中进行测试。b. 液体吸收池法: 对于低沸点液体样品和定量分析,要用固定密封液体池。制样时液体池倾斜放置,样品从下口注入,直至液体被充满为止,用聚四氟乙烯塞子依次堵塞池的入口和出口,进行测试。(3) 气态样品的制备:气态样品一般都灌注于气体池内进行测试。(4)特殊样品的制备—薄膜法a. 熔融法: 对熔点低,在熔融时不发生分解、升华和其它化学变化的物质,用熔融法制备。可将样品直接用红外灯或电吹风加热熔融后涂制成膜。b. 热压成膜法: 对于某些聚合物可把它们放在两块具有抛光面的金属块间加热,样品熔融后立即用油压机加压,冷却后揭下薄膜夹在夹具中直接测试。c. 溶液制膜法: 将试样溶解在低沸点的易挥发溶剂中,涂在盐片上,待溶剂挥发后成膜来测定。如果溶剂和样品不溶于水,使它们在水面上成膜也是可行的。比水重的溶剂在汞表面成膜。
GB/T 16773-2008 煤岩分析样品制备方法[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=]GBT 16773-2008 煤岩分析样品制备方法.PDF[/url] [color=#DC143C]标准制定者要求屏蔽此内容,涉及版权问题[/color]
各位大大,EPA中对同一类样品的制备和分析方法,是有多种的,请问下具体的分析这一类样品的时候,我们应该遵照哪一条呢?比如对于1,4,二氧杂环乙烷的分析,EPA有多个方法,如EPA8270,821,8260,1624,524.2.另外不同的规范下的方法是否有好差之分?谢谢。
98%,便于与纯化合物的标准进行对照。多组分试样应在测定前尽量预先用分馏、萃取、重结晶、区域熔融或色谱法进行分离提纯。(2) 试样中不应含有游离水。水本身有红外吸收,会严重干扰样品谱,而且还会侵蚀吸收池的盐窗。(3) 试样的浓度和测试厚度应选择适当,以使光谱图中的大多数吸收峰的透射比处于10%~80%范围内。包括控制浓度和压片的厚薄尺寸。2.制样方法(1) 固体样品的制备a.压片法:将1~2mg固体试样与200mg纯KBr研细混合,研磨到粒度小于2μm,在油压机上压成透明薄片,即可用于测定。b.糊状法:研细的固体粉末和石蜡油调成糊状,涂在两盐窗上,进行测试。此法可消除水峰的干扰。液体石蜡本身有红外吸收,此法不能用来研究饱和烷烃的红外吸收。(2) 液体样品的制备a. 液膜法: 对沸点较高的液体,直接滴在两块盐片之间,形成没有气泡的毛细厚度液膜,然后用夹具固定,放入仪器光路中进行测试。b. 液体吸收池法: 对于低沸点液体样品和定量分析,要用固定密封液体池。制样时液体池倾斜放置,样品从下口注入,直至液体被充满为止,用聚四氟乙烯塞子依次堵塞池的入口和出口,进行测试。(3) 气态样品的制备:气态样品一般都灌注于气体池内进行测试。(4)特殊样品的制备—薄膜法a. 熔融法: 对熔点低,在熔融时不发生分解、升华和其它化学变化的物质,用熔融法制备。可将样品直接用红外灯或电吹风加热熔融后涂制成膜。b. 热压成膜法: 对于某些聚合物可把它们放在两块具有抛光面的金属块间加热,样品熔融后立即用油压机加压,冷却后揭下薄膜夹在夹具中直接测试。c. 溶液制膜法: 将试样溶解在低沸点的易挥发溶剂中,涂在盐片上,待溶剂挥发后成膜来测定。如果溶剂和样品不溶于水,使它们在水面上成膜也是可行的。比水重的溶剂在汞表面成膜
[B][center]化学分析样品制备(1) 第一章 样品制备的基础知识(1)[/center][/B] 前言 分析化学研究的目的是得到所研究对象的有关信息,所分析的对象可以是气体、液体、或生物样品,要获得的信息如化学成分、结构信息、物理状态、表面性质或者在基因物质中的蛋白质的顺序。尽管动用了分析化学中所有的技术,也不好解决尽管是少数样品中的每一个信息。在很多情况下,当今仪器发展不到可以获得所有需要的信息,只能得到一部分必要的信息,一些分析的程序如图 1 所示。 取 样 样品储存 样品制备 样品分析 图 1 分析的程序 分析的第一步是取样,即从要分析的目的物采集样品,采集能代表原始整体样品所有特征的一部分物质,取样因目的物情况的不同而使用不同的方法,例如要分析湖水中的Ca+2,要知道情况的不同其浓度有所变化,深度不同、季节不同其浓度有所不同。第二个环节是对样品的存储,这是一个很重要的步骤,因为它在样品采集和分析之间存在时间的延迟,样品的存储必须保证其物理和化学特征保持不变,这样一来才能代表样品真正的分析结果。 样品准备就绪,下一步就是“样品制备”,许多样品不具备立刻进样分析的条件,例如分析肝中的农药,不可能直接拿肝做样品进行分析,必须把农药转移到溶液中才能分析。样品制备可能有不同的过程,这些过程如图 1-2 所列。但是样品制备的方法决定于要分析组分的基体和浓度大小,例如痕迹量分析比常量分析需要更苛刻的样品制备程序。 样品制备好之后,就可以选择适当的仪器进行分析了。要得到不同的信息就需要选择适当的仪器进行测量,例如,有机物需要用色谱进行分析,金属分析需要用原子光谱,DNA 序列测定需要用毛细管电泳,微观结构需要用电镜来测量。 样品均一化 样品进行萃取 样品进行富集 样品进行净化 样品进行分析 . 图 1-2 样品分析流程 (资料来源:“Sample Preparation Techniques in Analytical Chemistry“,Editor J. D. WINEFORDNER,2003)
[color=red]GB/T 30815-2014[/color] [font=宋体]表面化学分析[/font][font=宋体]分析样品的制备和安装方法指南[/font]
高聚物红外光谱分析的试样制备谢狄霖 陈忠 福建省医学院科学研究所,福州350001 厦门大学化学系,厦门360005摘 要:结合实际工作经验,介绍了高聚物试样红外光谱检测中常有的热压铸膜法,溶解铸膜法,热熔附着法,溶解附着法,热裂解法等试样制备技术。来源:维普
土壤样品的制备方法前 言 “十三五”国家将建成由35000个监测点位构成的土壤环境质量监测网络,其中普查点位20000个,每五年进行一次监测,风险点位15000个,一年进行一次监测。根据国务院办公厅印发的国办发〔2015〕56号《生态环境监测网络建设方案》精神:“健全生态环境监测法律法规及标准规范体系。研究制定环境监测条例、生态环境质量监测网络管理办法、生态环境监测信息发布管理规定等法规、规章。统一大气、地表水、地下水、土壤、海洋、生态、污染源、噪声、振动、辐射等监测布点、监测和评价技术标准规范,并根据工作需要及时修订完善。增强各部门生态环境监测数据的可比性,确保排污单位、各类监测机构的监测活动执行统一的技术标准规范。为了规范、指导和统一全国土壤环境监测工作,环保部成立了以监测司和中国环境监测总站领导组成的《土壤环境监测分析方法》领导小组,并由总站牵头,魏复盛院士领衔于2015年7月启动了《土壤环境监测分析方法》的编写工作。 相比水环境和环境空气质量监测,我国的土壤环境监测体系与质量控制指标严重滞后,在用的监测方法体系混乱,技术方法存在明显错误,所采用方法多来自农业(NY)、环境(HJ)、林业(LY)和国土(DD和DZ)等行业标准,不仅来自不同行业的同一方法存在差异,就环保部门自己不同版本的方法也各说各话,错误不少(详见http://bbs.instrument.com.cn/topic/5941369_1#floor_3)。从为广大环境监测工作者提供一本具有实用性、科学性和先进性重要参考书和工具书的思路出发,笔者现将所承担该书有关“土壤样品制备方法”的送审稿贴出来让大家先审。由于个人水平和理解有限,方法内容不当之处和疏漏在所难免,恳请版友批评指正和加入讨论。
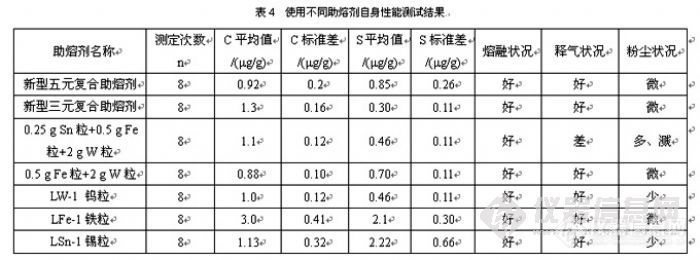
摘要:选用两个在C、S分析助熔剂专用粉末冶金生产线及其最佳工艺条件下制取的三元复合助熔剂和五元复合助熔剂,对HFIR法测定铁矿石类样品和生铸铁样品中C、S的分析性能分别进行研究并和其他助熔剂进行对比实验。结果表明,两种新型助熔剂均能全面显著地提高HFIR仪分析方法的技术水平:使仪器能进行各种样品中C、S的同时测定,其测定结果更准确、更精密、分析操作更简便、测定速度更快速。据此提出磁铁矿、赤铁矿、球团矿、烧结矿以及生铁、铸铁样品中C、S的HFIR的最佳分析方法。与此同时,进行不同助熔剂自身性能测试和对比实验,再次证实:选用的新型三元和新型五元复合助熔剂的优异分析性能源自于它们所具有的新材质特性,而这些新材质特性则是由独特的新制备工艺所赋予的。关键词:铁矿石中硫与碳;生铸铁中碳与硫;高频红外法;多元复合助熔剂;制备;性能近几年来,高频加热红外吸收(HFIR)法测定铁矿石以及烧结矿、球团矿中硫的研究取得重要进展:提出多元助熔剂分析方法,如0.2g样品+0.6g Fe屑+0.3gV2O5粉末拌匀的分析方法〔1〕和0.5gFe屑+0.05-0.25g样品+1.5gWSn粒的分析方法〔2〕,新发布实施的国标分析方法中采用0.9gFe屑+0.4g样品+0.4gFe屑+1.9gW粒的分析方法〔3〕,使得HFIR法已能够取代传统的化学分析法,成为铁矿石中新的分析方法。然而到目前为止,国内外都还没有HFIR C、S分析用三元或三元以上复合助熔剂可供使用,这些方法其实质都是使用一元的(或一元+二元的)助熔剂,都必须分别进行各种助熔剂的勺取,在电子天平上边称量边加入到装有样品的瓷坩埚中,其操作繁琐花费时间,必然影响仪器自动分析的连续性和降低分析速度,而且使得各种助熔剂的用量无法控制准确,更重要的是使这些外形各异、自身C.S含量不同、助熔剂性能不同的助熔剂,在样品上面按加入的先后分层或分堆堆积,从而造成被检测样品各部分的高频加热和C、S释放的不一致,成为影响分析结果准确度的重要因素。采用HFIR法测定生铁和铸铁中C、S〔4、5、6〕,因其称量少,且C、S又须同时测定,故亦存在着类似的样品难熔等主要问题。在自主设计并建成C、S分析助熔剂专用粉末冶金生产线后,我们一直在致力于新型助熔剂的研制工作〔7〕,现已成功开发并批量生产出多种HFIR C、S分析用三元或三元以上复合助熔剂。本文选用一个新型三元复合助熔剂和一个新型五元复合助熔剂,对HFIR法测定铁矿石类样品中 C、S的分析性能进行研究,并和不同助熔剂进行对比试验,结果表明:两个新型多元复合助熔剂均能全面显著地提高HFIR仪分析方法的技术水平。据此提出包括磁铁矿、赤铁矿、球团矿、烧结矿以及生铁、铸铁样品中C、S的HFIR的最佳方法。与此同时,进行不同助熔剂自身性能测试与对比实验,再次证实:选用的新型三元和五元复合助熔剂的优异分析性能是源自它们所具有的新材质特性,而这些新材质特性则是由独特的新制备工艺所赋予的。 1 实验部分1.1 C、S分析助熔剂生产工艺、设备和工具 C、S分析助熔剂专用粉末冶金生产线:自主设计和建成,其工艺流程和设备参见〔7〕容量可调取样勺:专利产品,使用前根据助熔剂用量调好并固定勺的容积,分析时可一勺按规定量把所用助熔剂取出并加入瓷坩埚中,省时又省工。1.2 分析仪器和试剂材料高频红外碳硫分析仪:选用国产经济型仪器,但具有程序加热和自动分析功能;根据使用需要,对分析气路进行一些必要的改进,并可与十万分之一电子天平配套使用,其测量灵敏度可达10-7%,检出限可优于10-5%,方法测定下限可低于10-4%〔8、9〕瓷坩埚:自产助熔剂:新型三元复合助熔剂、新型五元复合助熔剂、L-WA型W粒、L-Fe粒,L-Sn粒,均为自产。铁矿石样品:选用不同产地的铁矿石、磁铁矿、球团矿、烧结矿标样若干个;生铸铁样品选自不同产地的标样若干个;钢铁标样:超低碳硫纯铁YSBC20117C-2009和YSBC2012B-2008(上海钢研所)、碳素比色钢GBW01204a(上钢一厂),均作为对比验证样用。仪器气路吸水剂:自制新产品,吸水容量大且吸水后不潮解变形,即不改变气阻。1.3实验方法助熔剂制备工艺实验:通过大量重复试验,确定出每一种助熔剂的最佳生产工艺条件。本文选用的三元复合和五元复合助熔剂以及用于对比实验的W粒、Fe粒、Sn粒助熔剂都是在它们的最佳生产工艺制取,均具有下列新材质特性:1)均匀一致的外形;2)内部晶粒更细更匀;3)其C、S含量降至最低且均匀一致;4)多元复合剂的组分及其质量比按照分析方法要求而配制,其助熔性能是组合了各组分固有助熔性能而成为一种新的综合性能。HFIR分析方法实验首先在选择适宜且保持一直不变的高频加热条件和红外分析条件下,进行一定量(一勺)不同助熔剂对同一铁矿石类样品中C、S的HFIR分析性能实验,并同时进行一定量助熔剂下该样品不同称量的测定实验,以确定该样品的最佳助熔剂及其样品的称量范围。接着,选用C、S含量较高的标样进行校正分析并确定好C、S校正系数,又进行所用助熔剂的空白值校正分析并确定出空白校正值后,再进行不同C、S含量的铁矿石和生铸铁标样的连续测定,以观察分析方法的准确度、精密度和线性。最后以超低碳硫纯铁标样为校正样,进行不同助熔剂自身分析性能实验,进而探讨新型多元复合助熔剂具有优异分析性能的成因。2 结果和讨论2.1不同助熔剂HFIR测定铁矿石类样品中C、S的分析性能设定HFIR仪的加热功率为95%、加热时间为21秒、红外C、S分析时间均为40秒、截止电平均为2、系数均为1.0000、空白值均为0.0000、样品称量范围按其C、S含量高低并参考〔1-3〕选定。在这些条件保持不变的下,进行不同助熔剂在HFIR测定铁矿石类样品中C、S,包括样品熔融情况,CO2和SO2释放情况、产生的挥发粉尘量、分析结果及其精密度等分析性能的试验,结果如表1。[siz
红外光谱的样品制备第一部分液体 液样的制备是将少量样品涂于两片红外透明的窗片(KBr、NaCl等)之间。窗片的互相挤压形成一个样品薄层,样品的成分决定了选择哪种窗片。对于无水的样品,窗片材料是KBr。对于含水样品, KRS-5 较为适合。固体 固体样品对光谱学家提出挑战。样品的熔点为我们指出首先该考虑哪种技术。 对于熔点低于72。C的样品,用适当的溶剂将样品溶解,成膜于KBr窗片上是最先考虑的。如果因为基线不好或是溶解性差而不成功,可以考虑在两片KBr窗片内熔化成膜。如果这也不行,样品可进行KBr压片。 对于熔点高于72。C的样品,首选的技术是KBr压片。对于聚合物样品,成膜法是首选,接着是热熔法和压片法。 对于熔点未知的样品,结晶度的检测将会指明哪种技术将会成功。高结晶度的样品用KBr压片法较好,对于低结晶度的样品,成膜和热熔会得到更好的谱图。第二部分液体样品 液体样品的分析有多种方法。在本文中,我们主要探讨所使用的制样方法及一些有关的潜在问题。纯样品技术 分析液体样品的最常用方法就是将一滴液体夹在两片盐片中间,过程如下:将一滴样品滴于合适的盐片上,几秒钟后,将另外一块盐片合上,这样液体被夹在两块盐片之间,变成薄膜状。当然,选用的盐片要与分析的液体样品兼容。不含水的样品可采用KBr(32×5mm)盐片,含水样品则采用KRS-5盐片,这几种晶体材料的选用主要是根据它们在红外段的透光范围(优于4000-450cm-1)和稳定性。每次一个样品做好后,用带合适的溶剂的棉花清洗,然后在倒有甲醇的鹿皮或鸡皮上抛光。KBr盐片需要经常进行抛光,以维持其表面的光洁。由于KRS-5晶体有毒,所有只有当其表面被划伤或污染时才需要抛光,而且要求专业人员来完成。ATR技术 水平的单反射ATR主要是由一个ZnSe晶体的凹槽组成,尽管ZnSe晶体的截止频率为650-700CM-1,但它比其它宽频带的材料要更加耐用。 在样品分析好后,要用适当的溶剂将样品冲掉,再用棉花球擦洗干净,这种材料不需要经常抛光。潜在问题:但它最大的问题就是样品谱图的非线性,主要指峰位的位置和强度不满足Beer-Lambert法则:A = abc 这里, A =吸收值a =摩尔吸收系数 b = 光程c =浓度 Beer-Lambert定理主要是针对定量分析的,谱图检索是定量分析的一种类型,因为谱图检索是以吸收强度为基础的,透射实验一般是线性关系,可以用于定量分析;由于ATR的技术特点导致的,ATR实验一般不能用于定量分析。 最常见的导致非线性的原因是透过样品的光程不确定性。分辨率为2时,样品区红外光的聚焦点直径有6MM,如果此时样品区样品厚度薄厚不均或碰巧有气泡或,就会引起此处光程不同。这些因素将导致谱图在各个波段的吸收强度的不准确,换句话说,谱峰的强度比实际强度或者高或者低,从而降低谱图检索的质量,图1是纯3,4-二氯甲苯的红外吸收图,在808 cm-1波段处的吸收强度为0.39,而邻近870 cm-1处是0.24 (A808 cm-1/A870 cm-1 = 1.6)图2是同一个样品但是通过在晶体上做成一层薄厚不均的膜而得到的谱图,它的吸收率与上图已经有差别了,808 cm-1处的吸收率是0.76,而870 cm-1处却为0.62(A808 cm-1/A870 cm-1 = 1.2),与图1相比,已有25%的差距,这必然会导致谱图检索结果正确率的下降。将样品聚焦点直径为6mm时得到的图与聚焦点直径为3mm时得到的图相减,用差谱的结果来进行分析:由薄厚不均导致的非线性将会使差谱减不干净,有很大的残余峰,我们可以通过定期对晶体进行抛光来降低这种误差。 经常引起液体样品谱图的非线性的另一个原因是样品的厚度。液样太浓将会导致谱图的吸收太强,而多数红外仪器的检测器的线性响应范围是0到1.2个吸收单元,大于1.2时就会引起线性问题。有时非线性会使谱图中吸收峰的头部成平头状,在我们的实验室中只接受吸收单元1.2的谱图,图3也是上面提到的样品的谱图,但样品的厚度却远远大于前者。谱图中最强的吸收单元已经超过了30个吸收单元,808 cm-1处的吸收度为1.66,870 cm-1处为0.94(A808 cm-1/A870 cm-1 = 1.76),相比而言,产生了10%的误差,这种不同波段的吸收值的相对性的差异将会给谱图检索带来负面影响。第三部分成膜技术 涂膜技术是用在熔点低于72。C的样品和低结晶度的样品,比如象高聚物,涂膜法也可在其他方法失败后试用。涂膜的一般过程 先将样品溶于适当的溶剂中。然后将数滴溶液滴于惰性的基质上,溶液挥发后在基质上留下一层薄膜。如果惰性基质是红外透明的,可直接检测或将薄膜剥下检测。选择合适的溶液 选择溶液最主要的标准是容易挥发(除了最明显的一点,可溶解样品)。这意味着必须采用低沸点溶剂。蒸发溶剂所需的热量越少,样品所受的影响就越小。另外,溶剂越容易去除,残留的溶剂越少。以下列出的溶剂将首先考虑:氯仿(BP. 61.2° C),丙酮(BP. 56.2° C),三氯乙醇(BP. 151° C),邻二氯苯(BP. 180.5° C)和水(BP. 100° C)。在选择成膜技术时这五种溶剂适用于85%的样品。 纯溶液的光谱也应准备着作为参照。将溶剂的谱图与成膜样品的谱图作比较是判断是否有溶剂残留的一个好方法。每取用一次溶剂便将其参比谱图更新一下也是一个好习惯。选择基质 一般不将薄膜从基质上取下,基质和薄膜是一起放入光谱仪的。所以需要的是对红外透明的基质。除了溶剂是水采用KRS-5晶体外,一般最常用的基质是KBr晶体。如果决定将薄膜取下,玻璃将是不错的选择。成膜 经验告诉我们最好使用少量的稀溶液(3-5滴),多次在基质上形成薄膜,这将比用浓溶液形成的厚膜和大量的溶液一次成膜要好的多.这将使薄膜中的溶剂残留最少。有时,当你成膜的是晶体样品,谱图上会显示非常严重的散射和基线倾斜。这在单层成膜时经常发生,在多层成膜时也会出现。我们认为这是因为最先沉淀的晶体成为了形成大晶体的“晶核”,正是这些大晶体造成光的散射,使基线倾斜。在我们实验室为了防止这种问题的发生,我们经常在晶体的两面都涂上一层薄膜,有时在两块晶体的两面都涂上一层薄膜,一共形成四层膜。这个能解决绝大部分的散射问题。在蒸发溶剂时,使晶体上的溶液保持流动。这将帮助您得到厚度均匀的膜。我们经常将晶体放在一小片可反复使用的纸卡上(大约2”×3”),后不停的敲击纸卡的背面,使溶液保持流动,或者用移液管末端不停的搅拌搅拌,如果去除溶剂需要加热,而晶体又是水溶性物质,比如KBr,那你应该先加热卡片,去除其中含有的水汽。如果你不这样作,晶体的底部会吸水雾化,这将使你的谱图的基线倾斜。在我们的实验室,我们使用加热灯来慢慢清除水汽,如果是在一个较为潮湿的环境中,应该一直用灯加热 以去除环境中水汽的影响。注意采取预防措施,尤其是在使用易燃溶剂时。潜在问题 在成膜技术中最严重的两个问题是薄膜厚度不均匀和溶剂残留。薄膜的厚度不均将导致谱图的非线性。而在薄膜技术中应该时刻注意溶剂残留的问题。总是将得到的谱图与溶剂谱图的主峰作比较。如果结果显示有溶剂残留,有时可通过继续加热来去除溶剂。如果你不能确定某个特征峰是溶剂还是样品产生,那样品必须用另一种方法检测或使用另一种不会产生该特征峰的溶剂。另一个可能产生的问题是,某些样品在加热和有氧气的情况下易发生氧化。这将导致在1740 cm-1上有一个C=O 的小峰。有几种方法可以防止或减小这种氧化。在惰性气氛中蒸发溶剂,比如在氮气中,这样可以减少氧气的存在。或是减少加热量来化小这个问题。可能的话,你可以使用更低沸点的溶剂,或用真空泵来抽取溶剂。
红外光谱的样品制备第一部分液体液样的制备是将少量样品涂于两片红外透明的窗片(KBr、NaCl等)之间。窗片的互相挤压形成一个样品薄层,样品的成分决定了选择哪种窗片。对于无水的样品,窗片材料是KBr。对于含水样品, KRS-5 较为适合。固体固体样品对光谱学家提出挑战。样品的熔点为我们指出首先该考虑哪种技术。对于熔点低于72。C的样品,用适当的溶剂将样品溶解,成膜于KBr窗片上是最先考虑的。如果因为基线不好或是溶解性差而不成功,可以考虑在两片KBr窗片内熔化成膜。如果这也不行,样品可进行KBr压片。对于熔点高于72。C的样品,首选的技术是KBr压片。对于聚合物样品,成膜法是首选,接着是热熔法和压片法。对于熔点未知的样品,结晶度的检测将会指明哪种技术将会成功。高结晶度的样品用KBr压片法较好,对于低结晶度的样品,成膜和热熔会得到更好的谱图。第二部分液体样品液体样品的分析有多种方法。在本文中,我们主要探讨所使用的制样方法及一些有关的潜在问题。纯样品技术分析液体样品的最常用方法就是将一滴液体夹在两片盐片中间,过程如下:将一滴样品滴于合适的盐片上,几秒钟后,将另外一块盐片合上,这样液体被夹在两块盐片之间,变成薄膜状。当然,选用的盐片要与分析的液体样品兼容。不含水的样品可采用KBr(32×5mm)盐片,含水样品则采用KRS-5盐片,这几种晶体材料的选用主要是根据它们在红外段的透光范围(优于4000-450cm-1)和稳定性。每次一个样品做好后,用带合适的溶剂的棉花清洗,然后在倒有甲醇的鹿皮或鸡皮上抛光。KBr盐片需要经常进行抛光,以维持其表面的光洁。由于KRS-5晶体有毒,所有只有当其表面被划伤或污染时才需要抛光,而且要求专业人员来完成。ATR技术水平的单反射ATR主要是由一个ZnSe晶体的凹槽组成,尽管ZnSe晶体的截止频率为650-700CM-1,但它比其它宽频带的材料要更加耐用。在样品分析好后,要用适当的溶剂将样品冲掉,再用棉花球擦洗干净,这种材料不需要经常抛光。潜在问题:但它最大的问题就是样品谱图的非线性,主要指峰位的位置和强度不满足Beer-Lambert法则A = abc 这里, A =吸收值a =摩尔吸收系数b = 光程c =浓度Beer-Lambert定理主要是针对定量分析的,谱图检索是定量分析的一种类型,因为谱图检索是以吸收强度为基础的,透射实验一般是线性关系,可以用于定量分析;由于ATR的技术特点导致的,ATR实验一般不能用于定量分析。最常见的导致非线性的原因是透过样品的光程不确定性。分辨率为2时,样品区红外光的聚焦点直径有6MM,如果此时样品区样品厚度薄厚不均或碰巧有气泡或,就会引起此处光程不同。这些因素将导致谱图在各个波段的吸收强度的不准确,换句话说,谱峰的强度比实际强度或者高或者低,从而降低谱图检索的质量,图1是纯3,4-二氯甲苯的红外吸收图,在808 cm-1波段处的吸收强度为0.39,而邻近870 cm-1处是0.24 (A808 cm-1/A870 cm-1 = 1.6)图2是同一个样品但是通过在晶体上做成一层薄厚不均的膜而得到的谱图,它的吸收率与上图已经有差别了,808 cm-1处的吸收率是0.76,而870 cm-1处却为0.62(A808 cm-1/A870 cm-1 = 1.2),与图1相比,已有25%的差距,这必然会导致谱图检索结果正确率的下降。将样品聚焦点直径为6mm时得到的图与聚焦点直径为3mm时得到的图相减,用差谱的结果来进行分析:由薄厚不均导致的非线性将会使差谱减不干净,有很大的残余峰,我们可以通过定期对晶体进行抛光来降低这种误差。经常引起液体样品谱图的非线性的另一个原因是样品的厚度。液样太浓将会导致谱图的吸收太强,而多数红外仪器的检测器的线性响应范围是0到1.2个吸收单元,大于1.2时就会引起线性问题。有时非线性会使谱图中吸收峰的头部成平头状,在我们的实验室中只接受吸收单元1.2的谱图,图3也是上面提到的样品的谱图,但样品的厚度却远远大于前者。谱图中最强的吸收单元已经超过了30个吸收单元,808 cm-1处的吸收度为1.66,870 cm-1处为0.94(A808 cm-1/A870 cm-1 = 1.76),相比而言,产生了10%的误差,这种不同波段的吸收值的相对性的差异将会给谱图检索带来负面影响。第三部分成膜技术涂膜技术是用在熔点低于72。C的样品和低结晶度的样品,比如象高聚物,涂膜法也可在其他方法失败后试用。涂膜的一般过程先将样品溶于适当的溶剂中。然后将数滴溶液滴于惰性的基质上,溶液挥发后在基质上留下一层薄膜。如果惰性基质是红外透明的,可直接检测或将薄膜剥下检测。选择合适的溶液 选择溶液最主要的标准是容易挥发(除了最明显的一点,可溶解样品)。这意味着必须采用低沸点溶剂。蒸发溶剂所需的热量越少,样品所受的影响就越小。另外,溶剂越容易去除,残留的溶剂越少。以下列出的溶剂将首先考虑:氯仿(BP. 61.2° C),丙酮(BP. 56.2° C),三氯乙醇(BP. 151° C),邻二氯苯(BP. 180.5° C)和水(BP. 100° C)。在选择成膜技术时这五种溶剂适用于85%的样品。纯溶液的光谱也应准备着作为参照。将溶剂的谱图与成膜样品的谱图作比较是判断是否有溶剂残留的一个好方法。每取用一次溶剂便将其参比谱图更新一下也是一个好习惯。选择基质 一般不将薄膜从基质上取下,基质和薄膜是一起放入光谱仪的。所以需要的是对红外透明的基质。除了溶剂是水采用KRS-5晶体外,一般最常用的基质是KBr晶体。如果决定将薄膜取下,玻璃将是不错的选择。成膜 经验告诉我们最好使用少量的稀溶液(3-5滴),多次在基质上形成薄膜,这将比用浓溶液形成的厚膜和大量的溶液一次成膜要好的多.这将使薄膜中的溶剂残留最少。有时,当你成膜的是晶体样品,谱图上会显示非常严重的散射和基线倾斜。这在单层成膜时经常发生,在多层成膜时也会出现。我们认为这是因为最先沉淀的晶体成为了形成大晶体的“晶核”,正是这些大晶体造成光的散射,使基线倾斜。在我们实验室为了防止这种问题的发生,我们经常在晶体的两面都涂上一层薄膜,有时在两块晶体的两面都涂上一层薄膜,一共形成四层膜。这个能解决绝大部分的散射问题。在蒸发溶剂时,使晶体上的溶液保持流动。这将帮助您得到厚度均匀的膜。我们经常将晶体放在一小片可反复使用的纸卡上(大约2”×3”),后不停的敲击纸卡的背面,使溶液保持流动,或者用移液管末端不停的搅拌搅拌,如果去除溶剂需要加热,而晶体又是水溶性物质,比如KBr,那你应该先加热卡片,去除其中含有的水汽。如果你不这样作,晶体的底部会吸水雾化,这将使你的谱图的基线倾斜。在我们的实验室,我们使用加热灯来慢慢清除水汽,如果是在一个较为潮湿的环境中,应该一直用灯加热 以去除环境中水汽的影响。注意采取预防措施,尤其是在使用易燃溶剂时。潜在问题 在成膜技术中最严重的两个问题是薄膜厚度不均匀和溶剂残留。薄膜的厚度不均将导致谱图的非线性。而在薄膜技术中应该时刻注意溶剂残留的问题。总是将得到的谱图与溶剂谱图的主峰作比较。如果结果显示有溶剂残留,有时可通过继续加热来去除溶剂。如果你不能确定某个特征峰是溶剂还是样品产生,那样品必须用另一种方法检测或使用另一种不会产生该特征峰的溶剂。另一个可能产生的问题是,某些样品在加热和有氧气的情况下易发生氧化。这将导致在1740 cm-1上有一个C=O 的小峰。有几种方法可以防止或减小这种氧化。在惰性气氛中蒸发溶剂,比如在氮气中,这样可以减少氧气的存在。或是减少加热量来化小这个问题。可能的话,你可以使用更低沸点的溶剂,或用真空泵来抽取溶剂。
光谱法分析样品时,样品表面处理好坏将直接影响分析结果的准确度和精度,特别是对钢中氮分析,样品的制备尤为关键。那么怎么个关键法,大家讲讲!

石油地质行业中扫描电镜样品的制备方法 ,扫描电镜在石油地质行业中应用非常广泛。但目前国内、国外的扫描电镜用户在迚行样品制备的时候的经常会出现以下现象:样品内部的微小尺度结构在普通的手动研磨过程中会出现由于研磨所造成的表面的机械划痕、污染以及形变等各种损伤,很难得到其真实的形貌,很难观察到其内部的真实微区。在石油地质行业这种现象更加普遍,比如说这几年国家大力提倡的页岩气开发工作,因为页岩,泥岩,砂岩等样品涉及到其内部储气的孔隙通常都为纳米甚至是埃米级别才及地质样品本身的酥松性,遇水容易膨胀、变形和改变无形,在普通的手动机械抛光过程中,经常会出现堵塞孔隙的现象获得的电子扫描图像不能真实、直观地反映页岩的微观结构。空隙、裂缝、有机质及其它现象常会出现相同的图像效果,故当研究页岩的微观空隙结构时,通常的电镜方法难以凑效。所以现阶段国内外很多用户都会选择氩离子抛光装置来对于样品抛光,使用氩离子抛光处理样品表面后,电镜照片能更好的反应出页岩裂缝形态、空隙、有机质及其他矿物结晶甚至充填形态。氩离子抛光设备,依靠离子束轰击制备样品,从而能够获得传统加工方法难以达到的处理效果,是一个用于样品的截面制备及平面抛光的桌面型制样设备。可利用氩离子抛光设备进行抛光加工的材料种类十分广泛,包括由多元素组成的试样,以及具有不同的机械硬度、尺寸和物理特性的合金、半导体材料、聚合物和矿物等。如焊缝截面,集成电路焊点,多层薄膜截面,颗粒、纤维断面,复合材料、陶瓷、金属及合金、岩石矿物及其他无机非金属等各种材料的 SEM、EBSD 样品。氩离子光束抛光页岩样品表面后,结合扫描电镜、薄片岩相鉴定和 X-衍射仪等分析,可定量观察微孔隙结构,确定孔隙度,分析矿物成分。氩离子光束抛光制样技术在页岩研究中具有以下特点: (1) 氩离子光束抛光制样技术具有样品制备简便快捷,观察视域广、图像景深 大,放大倍数范围宽且连续可调,可迚行单组分细微结构的多方位观察,能对样品表面迚行多种信息综合分析等特点。(2)能够清楚地观察到岩石的主要空隙类型:粒间孔、微孔隙(包括粒内溶孔、杂基质微孔隙、微裂缝)、吼道类型(包括点状、片状和缩颈吼道)、测定出孔喉半径等参数和孔隙度。(3)岩样构造面、组分界面、矿物质、纳米级及其它更小的空隙、裂缝等,可较为方便地观察,可获得不同放大倍数较为优质的图像和照片。(4)可以判断有机质演化程度。http://ng1.17img.cn/bbsfiles/images/2016/11/201611161835_616613_0_3.png
1)易于重复,不同实验室内有经验的工作人员,依此方法可做出重复结果 (2)试剂用量少,以使样品测定时空白最低 (3)测定挥发性元素,如砷(As),汞(Hg),硒(Se),铅(Pb),锑(sb),锡(Sn),或组分如挥发油等在制样中不损失 (4)快速,满足于现代实验室的测定需要 (5)易于控制,实验室内不同人员依据操作方法可完成相同的任务 (6) 样品不被环境污染,制备的样品代表原始样品 (7)不污染环境,以利于保护实验室工作人员的健康.
各位XRF应用专家有没有使用重熔浇铸制备分析样品的,由于我们单位对检验结果的要求,压片法不能满足检验结果的准确度,玻璃融片法耗时成本高,不能满足我们的检验周期,想了解各位有没有使用这种方法及设备的。如果有请详细说明此种方法的效果。
斤方法应考虑的因素 样品中待测成分的分析方法往往很多,怎样选择最恰当的分析方法?一般地说,应该综合考虑下列各因素。 1.分析要求的准确度和精密度 不同分析方法的灵敏度、选择性、准确度、精密度各不相同。要根据生产和科研工作对分析结果要求的准确度和精密度来选择适当的分析方法。 2.分析方法的繁简和速度 不同分析方法操作步骤的繁简程度和所需时间及劳力各不相同,每样次分析的费用也不同。要根据待测样品的数目和要求取得分析结果的时间等来选择适当的分析方法,同一样品需要测定几种成分时,应尽可能选用同一份样品处理液同时测定该几种成分的方法。 3.样品的特性 各类样品中待测成分的形态和含量不同,可能存在的干扰物质及其含量不同,样品的溶解和待测成分的提取的难易程度也不相同。要根据样品的这些特征来选择制备待测液、定量某成分和消除干扰的适宜方法。 4.现有条件 分析工作一般在实验室进行,各级实验室的设备条件和技术条件也不相同,应根据具体条件来选择适当的分析方法。在具体情况下究竟选用哪一种方法,必须综合考虑上述各项因素,但首先必须了解各类方法的特点,如方法的精密度、准度、灵敏度等,以便加以比较。 资料来源:国家标准物质网资料中心
安捷伦样品制备及分析指南
液体液样的制备是将少量样品涂于两片红外透明的窗片(KBr、NaCl等)之间。窗片的互相挤压形成一个样品薄层,样品的成分决定了选择哪种窗片。对于无水的样品,窗片材料是KBr。对于含水样品, KRS-5 较为适合。固体固体样品对光谱学家提出挑战。样品的熔点为我们指出首先该考虑哪种技术。对于熔点低于72。C的样品,用适当的溶剂将样品溶解,成膜于KBr窗片上是最先考虑的。如果因为基线不好或是溶解性差而不成功,可以考虑在两片KBr窗片内熔化成膜。如果这也不行,样品可进行KBr压片。对于熔点高于72。C的样品,首选的技术是KBr压片。对于聚合物样品,成膜法是首选,接着是热熔法和压片法。
做岩矿分析样品分析的朋友,大家制备样品的情况怎么样,过程中混匀缩分都用什么方式,我们还是用掀角法混匀,四分法缩分,感觉有点落后,效率低下,质量也很难保证,大家交流一下,都是怎么做的.
各位朋友有去参加这个会的吗?到时候可以一起交流交流!2015年电子背散射衍射(EBSD)应用分析及样品制备技术研讨会 尊敬的客户:您好! 欧波同有限公司长期专注于微观纳米技术应用解决方案的研发与推广。经过数年发展,欧波同有限公司作为蔡司、牛津、GATAN公司战略合作伙伴,目前已经成为中国最大的微纳米显微方案供应商。为了推动EBSD技术及电子显微学的进步和发展,提高广大显微学工作者的学术及技术水平,促进显微学在物理学、材料学、生命科学、化学化工、环境、地学等领域的应用,欧波同有限公司、牛津仪器有限公司、GATAN公司三方联合于2015年7月24日举办2015年电子背散射衍射(EBSD)应用分析及样品制备技术研讨会。 本次会议将邀请电子显微学应用专家、EBSD应用专家、EBSD样品制备专家、SEM样品制备专家等进行相关领域的应用实例分析报告,同时将以欧波同有限公司专业化的DEMO实验中心为平台,为用户提供现场体验微纳米分析技术设备——蔡司电子显微镜,晶体学分析设备——牛津EBSD及EBSD(SEM)样品制备技术设备——GATAN ILION 697氩离子束抛光系统的一站式操作。欢迎您的到来! 会议主要议程安排 1、7月23日下午报到入住 2、7月24日 9:00~12:00专家报告 12:00~13:00午休 13:00~17:00 DEMO机考察及演示会议主要内容:1. 国内知名EBSD应用专家进行专题讲座,主要内容为EBSD在相关领域的应用实例;2. 欧波同公司扫描电镜最新技术以及仪器操作技巧介绍;3. 牛津公司介绍仪器的最新技术以及仪器设备维修与维护技巧;4. GATAN公司EBSD样品制备技术及结合EBSD的拓展应用介绍;5. 难处理扫描电镜样品制备的方法,包括镀层样品、电子半导体样品、高分子复合材料等截面样品制备,岩石矿物等孔隙样品制备等。6. DEMO机考察及演示(欧波同DEMO实验中心);会议地点:欧波同有限公司DEMO实验中心(北京市朝阳区高碑店乡西店村1106号源创空间大厦F16室)会议费用:欧波同提供会务费并赠送精美叫礼品,差旅费自理! 2015年电子背散射衍射(EBSD)应用分析及样品制备技术研讨会 回执函 单位名称: 地址: 邮编: 报名参加会议人员: 姓名 性别 职务 手机 备注 报名咨询联系人:刘丹 黄杨联系电话: 15140813412 18804252487Email邮箱:shchb02@163.com optonpo02@163.com[b
我们现在的直读光谱用来测定银、铜、铅等金属样品,样品制备都是用的普通车床。今年我们准备将生产的标准金锭也用直读光谱来测定,关于样品制备一直困扰我们。我们初步的设想是使用压片机进行制样,不知道是否合适?有没有更合适的样品制备方法?
分享一个红外光谱样品制备的一个非常好的ppt材料
菜鸟求教:用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析同一种物质时,如果样品制备方法不同,那么色谱条件是不是也就要不一样呢?
食品分析中,固体样品的平均样品制备,采用的方法是()。 A、先碎化,后混匀,用四分法制成平均样品 B、先搅后碎,再四分法制备成平均样品 C、用组织捣碎机捣碎后,取一部分成平均样品 D、直接混合,四分法制备平均样品
[align=center]农药残留分析样品的采样及样品制备规范[/align]摘要:本文按照标准要求,归集了在农产品产地采集样品时应规范采样单元的划分、样品的采集方法、采样量及样品的制备方法这里的农药残留分析样品采样是从种植业产地抽取产品的一部分作为其整体的代表性样本进行检测,再根据得到的质量数据和预先规定的判定规则判定“检查批”是否合格,因此,采样环节不可小视。下面就如何从“批”中采集样本,即采集什么样的“抽样方式”;应从“批”中抽取多少个单位产品,即应采集多大的样品量;对采样的样品如何制备来与大家共同探讨学习,如有不到之处,敬请指出…..[img=,427,372]http://ng1.17img.cn/bbsfiles/images/2018/08/201808170946349283_4003_1502196_3.png!w427x372.jpg[/img]一、采样单元的划分《农药残留分析样本的采样方法》(NY/T 789-2004)标准4.1条对产地样本的采样要求是:1.产地面积小于1hm[sup]2[/sup]时(按照NY/T 398规定划分抽样单元),以0.1~0.3hm[sup]2[/sup]划分一个抽样单元(即抽取1个代表性样品)。2.产地面积大于1 hm[sup]2[/sup]小于10hm[sup]2[/sup]时,每1 hm[sup]2[/sup]~3hm[sup]2[/sup]划分一个抽样单元,产地面积大于10hm[sup]2[/sup]时,每3 hm[sup]2[/sup]~5hm[sup]2[/sup]划分一个抽样单元。二、采样方法按照随机法、对角线法、五点法、Z形法、S形法、棋盘式法等多点采集。[img=,689,532]http://ng1.17img.cn/bbsfiles/images/2018/08/201808170951087607_8155_1502196_3.png!w689x532.jpg[/img]三、采样量1.蔬菜样品的采样量:①以0.1~0.3hm[sup]2[/sup]为一个抽样单元采集样品时,在抽样单元内选取5~20个植株,叶菜类整株采集;果实类在植株上、中、下各侧均匀采摘。②以1 hm[sup]2[/sup]~3hm[sup]2[/sup]为一个抽样单元采集样品时,叶菜类样品采样量至少为4个~12个个体,不少于3kg;果菜类(果皮可食)样品采样量至少为6个~12个个体,不少于3kg;果菜类(果皮不可食)样品采样量至少为4个~6个个体;茎菜类样品采样量至少为12个个体,不少于2kg。2.果品类样品的采样量:①以0.1~0.2hm[sup]2[/sup]为一个抽样单元采集样品时,在抽样单元内选取5~10株果树,每株果树纵向四分,从其中一份的上、中、下、内、外侧均匀采摘。②以1 hm[sup]2[/sup]~3hm[sup]2[/sup]为一个抽样单元采集样品时,柑橘类水果采样量至少为6个~12个个体,不少于3kg;梨果类水果采样量至少12个个体,不少于3kg;小水果和浆果采样量不少于3kg。3.食用菌类蔬菜的采样量:采集整个实体,至少12个个体,不少于1kg。四、样品的制备1.小体积蔬菜和水果,均匀混合后,按四分法缩分,用组织捣碎机或匀浆机处理后取250g-500g保存待测。[img=,497,497]http://ng1.17img.cn/bbsfiles/images/2018/08/201808171001148694_6918_1502196_3.jpg!w497x497.jpg[/img]2.大体积蔬菜和水果,切碎后,按四分法缩分,取600g-800g保存待测。五、抽样过程中易出现的问题1.采样单元布设不到位,对品种、来源忽视要求,未能对蔬菜的种类进行全覆盖采样,忽视了样本的代表性。2.对样品选择性比较随意,有时为了数量而忽视了样品质量。3.采样量小,因受检测样品用样量小,我们在抽样时往往不注意抽样单元,抽样量小,代表性不强。4.采样记录有漏记、漏签现象。例如:样本采样基数及采样数量、环境条件、人员签字等记录信息不全面。总结:要了解一个产地产品的质量,不可能对每个果实进行抽样检测分析,一般是通过从总体中抽取部分个体,根据对这部分个体的分析,对总体的性质作出估计判断,这就要求我们抽样人员在采集样品时做到布点准确、抽取样品质量完好、代表性强的样品,为农产品质量安全把好检验关。
介绍了XPS表征纤维样品的三种制备方法,并通过这三种制备方法对三类不同尺寸的纤维样品进行XPS分析及结果对比,进而展示三种制备方法的特点及适用范围。
内容包括XRD样品架的分类和使用优化方法,制样中的经验和特殊样品制备技巧,比如微区固体样品制备,微量样品的细节优化处理、不规则薄膜样品的制备,粉末样品颗粒度影响等,以保证高质量的实验数据。
食品样品的采取及制备首先明确的是食品分析的一般程序为:样品的采集、制备和保存,样品的预处理、成分分析、分析数据处理及分析报告的撰写。 那么什么是样品的采集呢?所谓采样就是从整批产品中抽取一定量具有代表性样品的过程。一. 采样的目的意义首先正确采样,必须遵守两个原则:第一,采集的样品要均匀,有代表性,能反应全部被测食品的组份,质量和卫生状况;第二,采样过程中要设法保持原有的理化指标,防止成分逸散或带入杂质。其次食品采样检验的目的在于检验式样感官性质上有无变化,食品的一般成分有无缺陷,加入的添加剂等外来物质是否符合国家的标准,食品的成分有无搀假现象,食品在生产运输和储藏过程中有无重金属,有害物质和各种微生物的污染以及有无变化和腐败现象。由于我们分析检验时采样很多,其检验结果又要代表整箱或整批食品的结果。所以样品的采集是我们检验分析中的重要环节的第一步,采取的样品必须代表全部被检测的物质,否则以后样品处理及检测计算结果无论如何严格准确也是没有任何价值。下面我们分别介绍对各种样品取样数量。所谓采样就是在原料或食品的成品中抽取具有一定代表性的样品。二、采样的数量与方法由于食品种类繁多,有罐头类食品,有乳制品、蛋制品和各种小食品(糖果,饼干类)等。另外食品的包装类型也很多,有散装的(比如粮食,食糖),还有袋装的(如食糖),桶装(蜂蜜)听装(罐头,饼干),木箱或纸盒装(禽,兔和水产品)和瓶装(酒和饮料类)等。食品采集的类型也不一样,有的是成品样,有的是半成品样品 ,有的还是原料类型的样品,尽管商品的种类不同,包装形式也不同,但是采取的样品一定要具有代表性,也就是说采取的样品要能代表整个班次的样品结果,对于各种食品取样方法中都有明确的取样数量和方法说明。我们举例如下:1)颗粒状样品(粮食,粉状食品)对于这些样品采样时应从某个角落,上中下各取一类,然后混合,用四分法得平均样品。下面我们对几个概念讲一下。上面我们提到,检样,原始样品,平均样品:检样—有整批食物的各个部分采取的少量样品称为检样。原始样品—把许多检样混在一起为原始样品。平均样品—原始样品经处理再抽取其中一部分作分析用的称平均样品2)半固体样品(如蜂蜜,稀奶油)用采样器从上中下分别取出检样混合后得平均样品。3)液体样品液体样品,先混合均匀,用吸法分层取样每层取500ml,装入瓶中混匀得平均样品。4)小包装的样品对于小包装的样品是连包装一起取(如罐头,奶粉)一般按生产班次取样,取样数为1/3000,尾数超过1000的方取1罐,但是每天每个品种取样数不得少于3罐。5)鱼、肉、果蔬等组成不均匀的样品根据我们检验的目的,我们可对各个部分(如肉,包括脂肪、肌肉部分、蔬菜包括根、茎、叶等)分别采样经过捣碎混合成为平均样品。如果分析水对鱼的污染程度,只取内脏即可.三.样品的制备与保存样品制备的目的,在于保证样品十分均匀,使我们在分析时候,取任何部分都能代表全部被测物质的成分,根据被测物的性质和检测要求,制备方法有下面几种1.样品的制备方法①摇动或搅拌(液体样品,浆体,悬浮液体) (用玻璃棒、电动搅拌器、电磁搅拌)②切细或搅碎 (固体样品)③研磨或用捣碎机对于带核、带骨头的样品,在制备前应该先取核、取骨、取皮,目前一般都用高速组织捣碎机进行样品的制备。2.保存采取的样品,为了防止其水分或挥发性成分散失以及其它待测成分含量的变化,应在短时间内进行分析,尽量做到当天样品当天分析。样品在保存过程中可能会有以下几种变化:①吸水或失水②霉变③细菌样品在保存时有几种变化(可能发生的变化)a)吸水或失水原来含水量高的易失水,反之则吸水,含水量高的易发生霉变,细菌繁殖快,保存样品用的容器有玻璃、塑料、金属等,原则上保存样品的容器不能同样品的主要成分发生化学反应。b)霉变特别是到新鲜的植物性样品,易发生霉变,当组织有损坏时更易发生褐变,因为组织受伤时,氧化酶发生作用,变成褐色,对于组织受伤的样品不易保存,应尽快分析。例如:茶叶采下来时,先脱活(杀青)即加热,脱去酶的活性。c)细菌为了防止细菌,最理想的方法是冷冻,样品的保存理想温度为-20℃,有的为了防止细菌污染可加防腐剂,例如甲醛,牛奶中可加甲醛作为防腐剂,但量不能加的过多,一般是1-2d/100ml牛奶。
各位专家我这现在新进一台荧光,在使用中遇到几个问题,希望大家能给以帮助1,对于不同的样品的样品制备的操作规程或者叫处理方法上,有没有国标或行标?上网找了许多都不是我们用的(我们主要做的是陶瓷及矿物原料),没有相关规程及标准,感觉样品制备,特别是熔样很难进行。2,对于未知样品,在没有标样的时候,怎样能进行比较准确的定量分析。希望各位不吝赐教,先谢过了
增材制造俗称3D打印,是近些年来飞速发展的新兴快速成型技术,在航空航天、电子电气等领域中发挥越来越重要的作用。同时,采用增材制造工艺制备的零件的测试和性能评估也越来越重要。金相样品制备和硬度测试是材料(包括增材制造材料)研发、制造、质量控制中常用的材料分析方法,且随着现代科技的进步趋于专业、高效和智能化。本报告将结合基础理论和实际应用案例,分享增材制造样品的高效率金相制样和硬度测试方法、设备、注意事项等,从而为参与者提供该领域制样和硬度测试解决方案的新思路。本报告是弗尔德仪器2021年围绕增材制造工艺开展的系列网络课程之一。会议开设300个免费名额,欢迎大家报名,欢迎分享![img=,690,210]https://ng1.17img.cn/bbsfiles/images/2021/06/202106021605181727_9546_2507958_3.png!w690x210.jpg[/img]报名链接:[url=https://www.instrument.com.cn/webinar/meetings/ZC2]点击打开链接[/url]







 我要推广仪器
我要推广仪器
 下载APP
下载APP