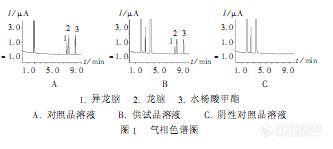
作者:付萍萍; 董秋香; 张月寒;(河北省保定市药品检验所;)摘要:目的建立醋酸氟轻松冰片乳膏的冰片含量测定方法。方法采用气相色谱法,以乙醇为溶剂,以水杨酸甲酯为内标,采用DM-5石英毛细柱、FID检测器,程序升温,100℃维持10min,再以20℃/min升至280℃,维持7min。结果冰片进样量在0.05002~1.0004μg范围内与峰面积线性关系良好(r=0.99996),平均回收率为99.74%(n=9)。结论该方法准确、灵敏,重现性好。谱图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207161105_377796_1606903_3.jpg

10,抽取5个版友);中奖名单:dyd3183621(注册ID:dyd3183621)莫名其妙(注册ID:moyueqiu)20071940xu(注册ID:20071940xu)yifan1117(注册ID:yifan1117)大川之子,纵横四海(注册ID:chuangu120)http://ng1.17img.cn/bbsfiles/images/2017/01/201701131506_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/01/201701131506_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================气相色谱法测定醋酸氟轻松冰片乳膏中冰片含量方法:GC基质:动物提取物应用编号:102052化合物:醋酸氟轻松,冰片固定相:DM-5色谱柱/前处理小柱:DM-5 30m x 0.32mm x 0.25um色谱条件:色谱柱:DM-5 30m×0.32mm×0.25 μm (Cat#:7231) 进样口温度:230℃ 尾吹气:氮气,流速:30mL/min 线速率:25cm/s,氢气流速30mL/min, 空气流速300mL/min 柱温:100℃保持10min,再以20℃/min升至280℃,保持7min 检测器温度:320℃ 载气:氮气(10:1) 进样量:1.0μL 检测器:FID检测器,温度 320℃文章出处:药物鉴定 2010,10(19):49-50关键字:醋酸氟轻松冰片乳膏,冰片,气相色谱法,DM-5谱图:摘要:目的建立醋酸氟轻松冰片乳膏的冰片含量测定方法.方法采用气相色谱法,以乙醇为溶剂,以水杨酸甲酯为内标,采用DM-5石英毛细柱、FID检测器,程序升温,100℃维持10min,再以20℃/min升至280℃,维持7min.结果冰片进样量在0.05002~1.0004μg范围内与峰面积线性关系良好(r=0.99996),平均回收率为99.74%(n=9).结论该方法准确、灵敏,重现性好.http://www.dikma.com.cn/Public/Uploads/images/1-10(1).jpg全文下载:气相色谱法测定醋酸氟轻松冰片乳膏中冰片含量
按2010版药典方法测定冰片含量时,用乙酸乙酯作溶剂,岛津2010气相色谱仪,进样1ul,峰面积总是不稳定。不知各位有没有遇到过这种情况
这几天做冰片的含量测定,峰总是分不开是什么情况
各位:谁有天然冰片的含量图谱,能不能给我两张呀,谢谢了。
冰片易挥发,结构如下: 请教:冰片测定一般采用GC法,如果采用紫外检测(DAD)的HPLC进行分析,色谱图会是怎样的呢?是根本就不会出峰还是会有小的峰,只是灵敏度太低?期待解答中……谢谢啦!

99.999% )文章出处:中国卫生检验杂志 2008, 18(6):1035-1036关键字:山药, 药材, 农药残留量, 气相色谱, GC, 电子捕获检测器谱图:摘要:目的:建立对中药、保健品、食品中有机氯类、拟除虫菊酯类等多种农药残留的气相色谱-电子捕获检测器(GC-ECD)测定方法。方法:选用DM-1毛细管柱,通过柱程序升温,对17种农药进行同时检测。结果:各种农药分别在10~200 ng/ml、20~400 ng/ml浓度范围内与峰面积呈良好的线性关系,检出限范围为0.092~3.25 pg。结论:本方法快速、简便、准确、具有良好的通用性。http://www.dikma.com.cn/Public/Uploads/images/29-10.JPG
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=63171]工业冰乙酸中甲酸含量的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[/url]工业冰乙酸中甲酸含量的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法
哪位朋友有丙酮氰醇含量测定的方法([url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法),可否给我上传一个,谢谢!
气相色谱测定冠心苏合丸中冰片的含量如何知道哪个峰时对照那样

安捷伦工作站标准曲线制作(以冰片中龙脑中含量测定为例)溶液的配制内标溶液:取水杨酸甲酯22.58mg至于50ml容量瓶中,加乙酸乙酯至刻度。摇匀即得。C内标=0.4516mg/ml对照品溶液:取龙脑对照品12.68mg至于10ml容量瓶中,加上述内标溶液至刻度,摇匀即得溶液I。精密量取溶液I 各2ml、3ml至于10ml容量瓶中用内标溶液稀释至刻度,得到0.2520mg/ml和0.3780mg/ml的龙脑溶液。样品溶液取冰片5.06mg至于10ml容量瓶中,加内标溶液至刻度。摇匀即得。C冰片 =0.5060mg/ml然后进样。得到图谱。下面主要讲讲标准曲线的制作。1、打开一张低浓度的图谱http://ng1.17img.cn/bbsfiles/images/2011/11/201111061959_328781_2088866_3.jpg(第一个峰位龙脑,第二个峰位内标化合物)选择calibration-------set up Quantitationhttp://ng1.17img.cn/bbsfiles/images/2011/11/201111062001_328782_2088866_3.jpg3设置定量参数http://ng1.17img.cn/bbsfiles/images/2011/11/201111062004_328783_2088866_3.jpg校正曲线标题(calibration title) 勾选 use RTEINT S使用RTE积分器下面为积分参数文件。你可以选择你编辑好的积分参数文件。这里我们不选就使用工作站默认的RTEint参数曲线拟合(curve fit ) 当然选线性拟合浓度单位( unit of concentration)内标化合物浓度(ISTD concentration)填好参数后http://ng1.17img.cn/bbsfiles/images/2011/11/201111062004_328784_2088866_3.jpg填好参数后点击ok如下图所示 进入编辑化合物界面http://ng1.17img.cn/bbsfiles/images/2011/11/201111161212_330835_2088866_3.jpg选择instert above 编辑化合物在总离子流图上右键双击得到总离子流图对于峰的色谱图。左右键同时按下选择用于定量的离子。我们这里内标化合物选择m/Z=152和120和92三个。内标化合物要勾选istd 并填入名字。点击save保存化合物。http://ng1.17img.cn/bbsfiles/images/2011/11/201111161213_330838_2088866_3.jpg注意编辑好的化合物会有一个黑色标记在相应的峰上http://ng1.17img.cn/bbsfiles/images/2011/11/201111161214_330840_2088866_3.jpg下面选择龙脑name填写龙脑 选择 M/Z 95和101的碎片http://ng1.17img.cn/bbsfiles/images/2011/11/201111161215_330841_2088866_3.jpg点击savehttp://ng1.17img.cn/bbsfiles/images/2011/11/201111161215_330842_2088866_3.jpg点击exit 退出http://ng1.17img.cn/bbsfiles/images/2011/11/201111161223_330849_2088866_3.jpg填写化合物浓度 校正水平填1 点击http://ng1.17img.cn/bbsfiles/images/2011/11/201111161225_330853_2088866_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/11/201111161228_330859_2088866_3.jpg调用另一张高浓度的图谱 先积分选择cali b ra tion u p d a te u p d a te anl e velhttp://ng1.17img.cn/bbsfiles/images/2011/11/201111161230_330861_2088866_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/11/201111161230_330862_2088866_3.jpg水平选择2 要输入第二针的浓度哦http://ng1.17img.cn/bbsfiles/images/2011/11/201111161227_330856_2088866_3.jpg再次进入编辑化合物界面 选择 find compoundhttp://ng1.17img.cn/bbsfiles/images/2011/11/201111161231_330863_2088866_3.jpg这时得到了校正曲线了。然后你可以载入样品数据,积分,选择定量 里面的计算。可以得出样品的含量。http://ng1.17img.cn/bbsfiles/images/2011/11/201111161234_330864_2088866_3.jpg另外校正曲线也可以导出到好奇怪老是提示 字符非法。我没有输入非法字符啊 !!!!????要不就是cookie过期传word版上来了。大家自己下载来看吧
冰片的薄层色谱一般为2个斑点,为什么有时候同样的展开剂跑出了1个斑点,是什么原因引起的?有点纳闷,薄层板是自己铺的
目的:建立一个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件同时分离测定环孢素A中乙醇及丙二醇的含量。方法:以GDX-101为固定相,柱长为2 m,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标。结果:乙醇及丙二醇进样量分别在2.0~6.0 μg,1.0~3.0 μg,其峰面积与浓度呈良好的线性关系,加样回收率分别为99.9%(RSD<0.8%,n=5),101.4%(RSD<1.1%,n=5),精密度良好。结论:此[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件可同时测定环孢素A中乙醇及丙二醇的含量,方法简便准确。关键词 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 乙醇 丙二醇 环孢素A山地明(环孢素A)为诺华制药有限公司的产品,是一种免疫抑制剂,用于器官移植和骨髓移植中的抑制排斥现象以及自身免疫疾病。厂方质量标准中乙醇及丙二醇的含量采用石英毛细管柱测定,此种色谱柱在国内使用不普及,我们经多次试验,摸索出一较好的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,适用于国内检测,即以GDX-101为固定相,柱长为2 m,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标,可同时分离测定环孢素A中乙醇及丙二醇的含量,改进后的方法,乙醇与正丙醇的分离度为3.1,丙二醇与正丙醇的分离度为5.0,符合中国药典1995年版中乙醇量度检查的分离度要求[1],操作简便,结果准确可靠。1 仪器与试药 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:SP-6890 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱:玻璃柱,长2 m,固定相为GDX-101。 乙醇、异丙醇、丙二醇均为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]纯,二甲基亚砜为色谱纯。 样品:环孢素A胶囊(山地明),由诺华公司提供,批号为187MFD0797;241MFD0797;166MFD0797;483MFD0797;477MFD0797。 标准贮备液及内标贮备液:精密称取[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]级的乙醇及丙二醇2.50及1.25 g分置50 mL容量瓶中,加二甲基亚砜至刻度,摇匀,作为标准贮备液;精密量取正丙醇5.0 mL置50 mL量瓶中,加二甲基亚砜至刻度,摇匀,作为内标贮备液。2 试验方法与结果2.1 色谱条件 采用GDX-101为固定相,柱长为2 m,氮气为载气,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,即初始为165 ℃,保持12 min,以40 ℃。min-1升至280 ℃,并保持20 min,检测器温度为280 ℃,进样量为2 μL。2.2 分离度试验 称取乙醇、丙二醇及正丙醇各50 mg置同一50 mL量瓶中,加二甲基亚砜至刻度,摇匀,进样2 μL,按上述色谱条件试验,记录色谱图,见图1-A,乙醇、丙二醇及正丙醇的保留时间分别为1.15,2.22,7.54 min,计算乙醇与正丙醇及丙二醇与正丙醇的分离度,其分离度分别为3.1和5.0。图1 分离度色谱(A)及样品测定(B)色谱图1.乙醇 2.正丙醇 3.丙二醇 4.二甲基亚砜2.3 线性范围及标准曲线 分别精密量取乙醇和丙二醇标准贮备液1.0,1.5,2.0,2.5,3.0 mL,分别置50 mL量瓶中,并分别加入内标贮备液1.0 mL,使乙醇终浓度为1.0,1.5,2.0,2.5,3.0 mg.mL-1,丙二醇的终浓度为0.5,0.75,1.0,1.25,1.5 mg.mL-1,分别进样2 μL,以乙醇及丙二醇的进样量为横坐标,以它们的峰面积与内标峰面积之比为纵坐标,分别进行线性回归,结果线性关系良好,乙醇、丙二醇回归方程分别为:A=8.935×103C+7.858×102 r=0.998 8A=8.086×103C-1.649×102 r=0.999 92.4 精密度试验 用乙醇与丙二醇浓度分别2.0及1.0 mg.mL-1的溶液,重复进样5次,结果乙醇与丙二醇的RSD分别为0.7%和1.0%,精密度良好。2.5 回收率试验 采用加样回收法,取已知乙醇与丙二醇含量的样品2粒,用二甲基亚砜溶解,置50 mL量瓶中,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,精密量取此溶液4.0,4.5,5.0,5.5,6.0 mL,分别加入乙醇与丙二醇的浓度分别为2.0 mg.mL-1及1.0 mg.mL-1的标准溶液6.0,5.5,5.0,4.5,4.0 mL,混匀,量取混匀后的溶液2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定这5份溶液的乙醇和丙二醇含量,计算回收率,乙醇的平均回收率为99.9%(RSD<0.8%,n=5),丙二醇的平均回收率为101.4%(RSD<1.1%,n=5)。2.6 样品的测定 取乙醇和丙二醇标准贮备液2.0 mL,内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为对照品溶液;取环孢素A胶囊2粒,置50 mL量瓶中,用二甲基亚砜溶解,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为样品溶液;分别量取对照品溶液和样品溶液各2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],按上述色谱条件测定,以内标法计算含量,即得;见图1-B。2.7 对比试验结果 取环孢素A样品5批,用改进后的方法测定样品中乙醇和丙二醇的含量,与厂方测定数据相比,结果基本吻合,见表1。表1 乙醇和丙二醇对比试验结果(%) 批号 本法结果 厂方测定数据 乙醇 丙二醇 乙醇 丙二醇 187MFD0797 101.0 106.3 100.5 105.0 241MFD0797 99.2 99.2 100.6 100.6 166MFD0797 101.7 102.7 101.3 103.0 483MFD0797 98.8 96.8 99.3 97.2 477MFD0797 99.1 98.1 98.9 97.7 3 讨论3.1 本法与原厂方方法相比,方法更为简便,条件普及,有利于对样品质量的控制。3.2 原厂方标准在测定乙醇含量时,以正丁醇为溶剂,由于正丁醇的保留时间与丙二醇过于接近,分离度达不到要求,本法采用二甲基亚砜为溶剂,不影响样品的溶解,同时使丙二醇与二甲基亚砜的分离度符合定量分析的要求。3.3 曾用固定相为GDX-401的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]柱进行检测,乙醇与正丙醇得到完全分离,但丙二醇与溶剂峰重叠,分离度达不到要求。3.4 采用程序升温,可使溶剂出峰时间加快,缩短分析时间。王俊秋(北京市药品检验所 北京 100035)庞青云(北京市药品检验所 北京 100035)余立(北京市药品检验所 北京 100035)参考文献1,中国药典.1995.二部:附录44
请问肉豆蔻酸异丙酯(BP)的含量测定(GC)的色谱柱,用哪种?

10,抽取5个版友);中奖名单:大川之子,纵横四海(注册ID:chuangu120)20071940xu(注册ID:20071940xu)yifan1117(注册ID:yifan1117)m3071659(注册ID:m3071659)夏天的雪(注册ID:bingwang228)http://ng1.17img.cn/bbsfiles/images/2017/02/201702101519_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/02/201702101519_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================气相色谱法测定小金片中麝香酮含量方法:GC基质:药品应用编号:102823化合物:麝香酮,小金片固定相:DM-17色谱柱/前处理小柱:DM-17 30m x 0.25mm x 0.25um色谱条件:色谱柱:DM-17 30m×0.25mm×0.25 μm (Cat#:7421) 进样口温度:230℃ 柱温:200℃ 检测器温度:250℃ 分流比:30:1 载气:氮气 进样量:0.2μL 检测器:FID检测器文章出处:中国药业 2008,8(17):38-39关键字:气相色谱法,麝香酮,小金片,含量测定,DM-17谱图:摘要 目的气相色谱(GC)法测定小金片中麝香酮含量.方法以乙醇为溶剂提取小金片中的麝香酮,采用DM-17毛细管色谱柱(30.0m×0.25m m,0.25μm),FID检测,柱温200℃,进样口温度230℃,检测器温度为250℃,载气为N2.结果麝香酮进样量在17.856~178.56ng范围内与峰面积线性关系良好,r=0.9993;平均回收率为95.7%,RSD为0.89%(n=6).结论方法准确、灵敏,重现性好,可用于小金片的质量控制。http://www.dikma.com.cn/Public/Uploads/images/83-10.jpg
【作者】 郭宏; 高金波; 邱树勇;【Author】 Guo Hong, Gao Jinbo, Qiu Shuyong (Jiamusi University, Jiamusi 154002)【机构】 佳木斯大学佳木斯!154002;【摘要】 目的:建立高效液相色谱法测定恶丙嗪片含量的方法.方法:色谱柱: DiamonsilC18(200 × 4.6mm, 5μm),流动相:甲醇水(80: 20),检测波长:285nm.结果:恶丙嗪线性范围5~45μg/ml,回归方程为C=0. 0624A+45. 229r=0.9999,回收率为99.86%,日内精密度为RSD=0.92%,日间精密度为RSD=1.64%结论:方法准确、快速、简便.
心力丸是本所科研成果,药厂注册品种,国家中药保护品种,质量标准收载于卫生部中药成方制剂第十七册(WS3—B—3150—98)。本品具温阳益气、活血化瘀功能,用于心阳不振、气滞血瘀所致胸痹心痛、胸闷气短、心悸怔忡等证及冠心病、心绞痛。该制剂是由麝香、人参、附子、红花、人工牛黄、蟾酥等中药制成的浓缩丸,现国家规定处方中麝香以人工麝香等量替代使用,因此,人工麝香中主要有效成分麝香酮的含量[1],对控制该产品质量和保证临床用药安全有效具有重要意义。已有对六神丸等制剂中麝香酮进行测定的文献[2-5]报道。本文用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定心力丸麝香酮的含量,方法简便、准确,可以有效控制该产品质量。1 仪器和试剂1.1 仪器岛津GC17A[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],FID 检测器,N2000数据工作站(浙江智达) METTLER AE240电子分析天平 AS5150A超声波仪。1.2 试剂、试药麝香酮对照品(中国药品生物制品检定所,批号:071920006,含量测定用,质量分数99.8%) 心力丸(广东省药物研究所制药厂) 无水乙醇、乙醚等试剂均为分析纯 压缩空气、H2、N2均为高纯度的气体(广州气体厂,质量分数达99.9%以上)。2 方法与结果2.1 色谱条件毛细管柱:SPBTM1 FUSED SILICA Capillary Column(30 m×0.32 mm×1.0 μm) 柱温:200 ℃ 进样口温度:210 ℃ 检测器温度:225 ℃ 载气:N2 流速:2 mL/min 柱前压:80 kPa 空气:300 mL 氢气:30 mL 尾吹:30 mL 分流:50∶1 进样量:1 μL。在上述条件下,供试品溶液中麝香酮色谱峰能达到基线分离,供试品与邻峰的分离度大于2.0,峰形对称,人工麝香阴性供试品无干扰,方法具有专属性,按麝香酮计算理论塔板数不低于4 500。色谱图见图1-图3。图1 麝香酮对照品的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(M.麝香酮)(略)Figure 1 Gas chromatograms of muscone图2 心力丸供试品的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(M.麝香酮)(略)Figure 2 Gas chromatograms of Xinli pills图3 人工麝香阴性[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(略)Figure 3 Gas chromatograms of negative sample2.2 线性关系考察精密称取麝香酮对照品0.151 94 g,置100 mL量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀 再分别精密量取0.5、1、2、4、5、6、8、10 mL置10 mL量瓶中,加无水乙醇至刻度,摇匀,精密吸取1 μL进样,测定峰面积,以对照品质量浓度(ρ)为横坐标,峰面积(A)为纵坐标,求得回归方程为A=291382. 2ρ+18117.0,r=0.999 6,麝香酮线性范围为0.076~1.5 mgmL-1。2.3 供试品溶液制备取本品,研细,精密称取3.5 g,置具塞锥形瓶中,加入乙醚30 mL,密塞,冷浸12 h,滤过,残渣和具皿用少量乙醚洗涤数次,合并乙醚液,挥去乙醚,残渣加无水乙醇使溶解并定容至5 mL,摇匀,用0. 45 μm滤膜滤过,取续滤液,即得。2.4 测定方法精密吸取供试品溶液和对照品溶液1 μL,分别注入[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]中,记录色谱图,以外标法计算供试品中麝香酮含量。2.5 稳定性试验取同一供试品溶液制备后室温放置,分别在0、1、2、4、6、8、10 h进样测定,每次进样1 μL,测得麝香酮平均含量490.4 μg/g,RSD为0.29%。结果表明供试品在10 h内测定结果稳定。2.6 精密度试验取麝香酮对照品溶液(0.759 7 mg/mL)重复测定6次,测得麝香酮峰面积的RSD值为0.15%。2.7 重现性试验取同一批供试品,平行6次精密称取,按“2.3”项下方法制备供试品溶液,依“2.4”项测定,计算供试品中麝香酮含量,结果其RSD值为1.4%,表明方法重现性好。2.8 回收率试验精密称取已知含量的心力丸(麝香酮含量495. 0 μg/g)共9份,置具塞锥形瓶中 取高(1.5194 mg/mL)、中(0.759 7 mg/mL)、低(0.075 97 mg/mL)3个浓度的对照品溶液各3份,每份2 mL,置蒸发皿中,通风柜中挥干乙醇,残渣用适量乙醚溶解,移入称取有供试品的具塞锥形瓶,用乙醚洗涤具皿数次,将洗涤液并入锥形瓶,乙醚用量为30 mL,其余按“2.3”方法制备,按“2.4”方法测定,结果见表1。2.9 样品测定取本品6批,按“2.3”项下方法制备供试品溶液,按“2.4”法进行测定,结果见表2。表1 麝香酮回收率试验结果(略)Table 1 Recovery test of muscone表2 样品测定结果(略)Table 2 Assay test of samples3 讨论3.1 提取方法的选择分别考察了超声提取法和浸渍法,结果见表3。结果表明:用乙醚作溶剂的2种提取方法有偏差(相对偏差为3.1%),浸渍法提取比超声法提取损失少,或者说提取较完全 在产品含量测定时采用浸渍法提取,提取适宜时间为12 h 在生产过程中的产品质量检测则可采用超声提取方法。3.2 色谱柱的选择曾采用强极性毛细管柱(SHIMADZU CBP20睲25025)进行试验,结果麝香酮很难达基线分离,且被测组分峰有拖尾现象。表3 两种提取方法麝香酮含量测定结果(略)Table 3 Muscone contents from two batches of extractions【参考文献】[1] 唐洪梅,黄樱华,李得堂,等.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定人工麝香中麝香酮的含量[J].中国实验方剂学杂志,2007,13(9):4-6.[2] 林巧玲,卓婷.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定保婴散中麝香酮的含量[J].广东药学院学报, 2006,22(3):257-258.[3] 袁劲松,汤翠娥.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定紫金胶囊中麝香酮含量[J].中国医院药学杂志, 2003,23(2):89-91.[4] 邹巧根,苏建俊.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定麝香保心丸中麝香酮的含量[J].中国中药杂志, 1994,19(7):418-420.[5] 王强,毕开顺,陈金泉,等.六神丸中麝香酮含量的GC法测定[J].中国药科大学学报, 1995,25(6):87-89.
求助:GB/T 5009.204-2005 食品中丙稀酰胺含量的测定方法 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱(GC-MS)法各位老大!帮帮忙,本人急需此标准。
[color=#444444]我想知道[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]如何测定回收液中乙醇和丙酮含量,我主要想用FID检测器,求各位大侠指点,如果有详细点的步骤最好了,我早上进样试了试貌似两个峰重合了。如果用内标法的话可以嘛?用什么做内标比较好,求具体过程[/color]
[color=#444444]现在打算用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定样品中各成分的含量,但由于对照品有些买不到,样品里面成分也很多,不可能买齐所有对照品,买了几个对照品,我想问问用内标法和一两个对照品能不能测出其他成分的含量?[/color]
[align=center][b][size=18px]运用气相色谱内标法测定食品中甜蜜素含量 [/size][/b] [/align] 甜蜜素化学名称为环己基氨基磺酸钠,于1937年发现,1950年开始生产应用。它是由环己胺和氨基磺酸或三氧化硫反应后用NaOH处理,再重结晶制得的一种白色结晶粉末,甜度为蔗糖的30~80倍。风味较自然,后苦不明显,热稳定性高,是不被人体吸收的低热能甜味剂。1969年曾因其致畸性的报道而被世界各国禁用,后来由于大量试验表明它并无致畸、致癌等作用,许多国家重又许可使用。我国于1987年开始应用甜蜜素,它是目前我国食品行业中应用蕞多的一种甜味剂。甜蜜素含量检测目前有气相色谱检测方法、液相色谱检测方法和液相色谱-质谱/质谱法等,但应用最为广泛的方法是依据GB/T5009.97-2016《食品中环己基氨基磺酸钠的测定》标准中的气相色谱仪分析法。色谱分析技术人员采用在提取溶剂正己烷中加入两种内标物质(甲苯和乙酸正丁酯)对甜蜜素经过衍生后的产物进行定量,具有快捷,准确等特点,受到普遍欢迎。1.试验部分1.1原理在硫酸介质中甜蜜素与亚硝酸钠反应,生成环己醇亚硝酸酯,利用气相色谱法进行定性和定量。1.2试剂(所用试剂不做说明皆为分析纯,水为蒸馏水)1.2.1甜蜜素储备溶液:称取1.0000g甜蜜素(含量≥99.0%),加水溶解并定容至100mL,此溶液浓度为10.00mg/mL,为储备液。置于4℃的冰箱中。本次试验溶液浓度为:10.320mg/mL1.2.2甜蜜素标准使用溶液:取1.2.1储备液10mL,加水定容至100mL,为使用液,浓度为1.0000mg/mL。本次试验使用溶液浓度为:1.0320mg/mL1.2.3100g/L硫酸溶液:称取50g浓硫酸,用水定容至500mL。1.2.450g/L亚硝酸钠溶液:称取25g亚硝酸钠,用水定容至500mL。1.2.5正己烷1.2.6甲苯(内标)1.2.7乙酸丁酯(内标)1.2.8氯化钠1.3仪器1.3.1气相色谱仪GC-2020(带FID检测器、毛细管进样口、N2000色谱工作站)1.3.2DB-5毛细管柱(30m×0.32mm×0.25μm)或其它类型毛细管柱,如DB-1、DB-17011.3.3离心机1.3.410μL微量进样器1.4仪器操作条件1.4.1检测器:200℃1.4.2汽化室:180℃1.4.3柱温:70℃1.4.4载气流速(压力):100kPa1.4.5氢气流速(压力):50kPa1.4.6空气流速(压力):60kPa1.5样品前处理1.5.1内标溶液的配置:在正己烷中加入一定量的内标(乙酸丁酯和甲苯)1,加入的量以出色谱峰合适为宜。一般为500mL正己烷中加入内标100μL~200μL。本次实验乙酸丁酯的浓度为0.1344mg/mL(称量0.0672g乙酸丁酯于装有10mL左右正己烷的25mL容量瓶中,再将溶液转移至500mL容量瓶中并定容至刻度线,摇匀,备用。1.5.2标准溶液前处理:吸取1.2.2标准溶液10mL于50mL比色管中,加10mL水,摇匀,置于冰浴中。加入5mL50g/L亚硝酸钠溶液,5mL100g/L硫酸溶液,在冰浴中放置30min,并时常摇动,然后准确加入10mL1.5.1溶剂,5g氯化钠,摇匀后振摇80次。静置分层。吸出正己烷层于10mL带塞离心管中进行离心分层(如有机溶剂和水相很快就有分层或只要进样针能吸出提取的溶剂,就可以不需离心分离这一步骤),吸取正己烷层1μL注入色谱仪进行分析。算出校正因子2。1.5.3液体样品前处理:称取试样20.0g于50mL带塞比色管中,加10mL水。摇匀,置于冰浴中。处理过程同标准溶液前处理1.5.2。吸取正己烷层试样1μL注入色谱仪进行分析。1.5.4固体样品前处理:称取已磨碎(剪碎)试样2.0~10.0g(根据样品中甜蜜素含量而定称取质量,使甜蜜素的量在1~10mg之间)于50mL带塞比色管中,加10mL水。一些样品不易溶解,如蜜饯类、山楂等,置于水浴锅中煮沸15min左右,冷却至60℃以下,置于冰浴中。处理过程同标准溶液前处理1.5.2。吸取正己烷层试样1μL注入色谱仪进行分析。2结果与讨论2.1.1分析方法的线性相关性的测定分别吸取1.2.1标准溶液1mL、3mL、5mL、10mL、20mL于5个100mL容量瓶中,并定容至刻度,配成浓度分别为0.1032mg/mL、0.3096mg/mL、0.5160mg/mL、1.0320mg/mL、2.0640mg/mL的标准溶液。分别吸取以上标准溶液10mL于5支50mL比色管中,按1.5.2方法进行处理。每个标准点分别进样5次,每次进样1μL,以甜蜜素与内标物的质量比为横坐标,甜蜜素与内标物的峰面积比(取五次进样平均值)为纵坐标绘制标准曲线,得线性方程为y=0.43991x-0.00088,其线性相关系数为0.99999。2.1.2分析方法的准确度的测定从市场上购买一种不含甜蜜素的饮料(样品名称:鲜橙多橙汁饮料生产单位:昆山统一企业食品有限公司净含量:2L生产日期:20060307),称取5份一定量的试样,用移液管分别加入1.2.2标准溶液1、3、5、10、15mL,按上述气相色谱操作条件测定甜蜜素回收率,结果都在99.7%~101.1%之间。2.1.3分析方法的精密度的测定从市场上购买一种含有甜蜜素的饮料(样品名称:鲜橙多生产单位:康裕食品有限公司净含量:1.5L生产日期:20060415),从同一产品中称取五个试样。按1.5.2方法进行处理后,按上述气相色谱操作条件测定甜蜜素含量,得到标准偏差为0.0027,变异系数(RSD)为0.55%。3结论综上所述,本方法简便、快速、准确,具有较高的准确度、精密度和回收率,线性关系好,是一种非常可行的分析方法。注:1用甲苯和乙酸丁酯做内标都可以,可同时加入两种内标(选其中一种作为参照计算),也可以只加其中一种。本次试验的数据是以乙酸丁酯为内标进行计算而获得的。2甜蜜素用本方法衍生处理后,在毛细管柱上会出两个峰,一般是刚开始前面的大,后面的很小,但时间长了,后面的峰越来越大,相应的,前面的峰越来越小直至消失,但两个峰面积和是不变的(48小时之内)。计算校正因子时,用甜蜜素衍生产物的两个峰的面积和计算。检测样品时,两个峰可单独识别并采用同一个校正因子计算,结果相加,也可以使用工作站将峰面积相加,再计算含量。一批样品检测,样品和标准品使用同一浓度的内标溶剂,在计算校正因子时,可将内标物的量假定为1(任一定值皆可),使用非常方便,不需知道加入的内标物的质量。否则,要称量内标物的质量,[font=FZSSK--GBK1-0]精确[/font]至0.0001g,再定容至一定体积。算出内标物浓度,再进行校正因子的计算。
[color=#444444]我最近做性能测试要测试醇水混合物以及酚水混合物各组分含量的确定,查看资料要用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。以前没有接触过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],假如我现在用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定乙醇与水混合物中乙醇和水的含量,应该选用哪种类型的柱子?如果我用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定苯酚和水混合物中苯酚和水的含量那应该用哪种柱子?这两种测试的柱子是不是可以通用?希望各位不吝赐教,在下感激不尽![/color]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152824]氧弹燃烧-[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用法测定废旧冰箱保温材料中CFC-11的含量[/url]摘 要:介绍了一种测定废旧冰箱保温材料(聚氨酯硬泡)中cFc~11含量的方法。聚氨酯硬泡中的cFc一11测定包含两部分:一是通过氧弹燃烧一[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定材料燃烧后的氯离子含量,另一部分是用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]一质谱联用法测定材料燃烧后CFC—l1的含量。
请问各位大侠,我每次做这个鉴别做出来的都是两个斑点,对照品也是两个斑点,对应位置,是正常的吗?对照品只有一种成分,就是冰片对照品,为什么会有两个斑点?有做过同样实验的指点一下咯~谢谢啦!
摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.1实验部分:1.1仪器和试剂电子分析天平(万分之一);GC7800气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mmx30mx0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4 1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2测定原理 试样中加适量内标物并用少许丙酮(4 1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3测定条件以样品中各组分是否完全分离开为依据而确认测定条件: 气化温度:280℃检测温度:320℃载气:氮气:纯度≥99.99%,变色硅胶 5A分子筛除水、除油,柱前压力为60Kpa(30℃);氢气:纯度≥99.99%,变色硅胶 5A分子筛除水、除油,柱前压力为65Kpa(30℃);空气:变色硅胶 5A分子筛除水、除油,柱前压力为55Kpa(30℃);柱温采用程序升温:初温30℃,恒温3min,以10℃/mm升温速率升至140℃,再以50℃/min升温速率升至260℃保持4min.分流比:45:1进样量:0.2ul1.4相对校正因子的测定1.4.1标准样品的配制于20ml样品瓶中分别称取0.03g(精确至0.0001g)的醋梭乙烯酯VAM、丙烯酸丁酯Bt等待测单体的色谱纯品和内标物环已酮,加入纺2mL丙酮(4 1)稀释,用力摇匀3min.(注意:每次衡量后应立即将样品瓶盖紧,以防止样品挥发损失。) 1.4相对校正因子的测定待仪器稳定后,吸取0.2uL标准样品注入色谱仪,记录色谱图和色谱数据……。典型合成树脂乳液的色谱图各单体及内标物的相对保留时间分别是:VAM1.771’;MMA3.078’;BA5.498’;St5.683’;2-EHA8.495’;内标环已酮6.218’1.4.3相对校正因子的计算醋酸乙烯酯、丙烯酸丁酯、苯乙烯等各需待测的残余单体对环已酮的相对校正因子Fi按下式计算:式中:mi——待测残余单体各自的质量;gAs——内标物环已酮的峰面积;ms——内标物环已酮的质量;gAi——待没残余单体各自的峰面积。 1.5连续平行测得待测残余单体各自对环已酮(内标物)的相对校正因子Fi的平等偏差应小于0.05.1.5加标回收率试验为了证明测定结果的可靠性,称取一定量的各待测单体纯品配制一已知尝试溶液,按上述分析方法测定各溶液的浓度,计算各自的回收率,结果见表(1) 1.6样品的测定将样品搅拌均匀后,在20mL样品瓶中准确称取2g左右和一小滴(用1mL注射器)内标物(约0.001g)环已酮于样品瓶中,加入适量(2~3mL)的丙酮(4 1)稀释剂,立即加盖瓶塞,充分摇匀2min.在相同于测定校正因子的分析条件下,用1ul微量进样器取0.2ul该试液注入色谱仪,并记录色谱图和数据,根据待测残余单体各自对内标物的保留时间进行定性。 1.7结果的计算待测残余单体各自的质量分数按下式计算:式中:Fi——待没单体各自对内标物的相对校正因子;Ms——内标物的质量,gAi——试样中待测残余单体各自的峰存积;Mi——待没试样的质量,gAs——内标物的峰面积取平行测定2次结果的算术平均值作为试样中各待测单体的测定结果。 1.8重现性同一操作作者两次测定结果的相对偏差小于0.5%2.结果与讨论(1)该分析方法选用中等极性小口径耐高温的毛细柱,进样量不能大于0.2ul,且分流比要足够大。由于各残余单体的沸点相对较高,所以我们选用耐高温的毛细柱,且该柱对酯类有较佳的分离效果。 (2)汽化室内须配有石英玻璃衬管,内填适量经处理的玻璃棉,以防止残留物进入毛细柱,并要勤换衬管为妥。 (3)因各单体对内标物的相对响应值不同,的以在配制标准溶液时,称取的各单体质量不一定都是0.03g,原则是尽量使各单体的峰面积和内标物的峰面积之比值接近于1。 (4)某些样品经丙酮稀释旋转一段时间后会出现破乳分层,做样品时须取其上层清液进色谱仪分析。 (5)醋梭乙烯酯VAM存在水的现象。因此在做其回收率时溶剂用脱水分析砘丙酮这样能有效控制VAM水解。但测试样时,溶剂为丙酮水溶液(4 1),这是由于有些样品溶于丙酮(脱水)时会产生胶化现象。
我们的气相色谱测定的样品都是溶解在溶剂中的,但是要怎么测定某个气体样品的含量呢?谢谢!
合成树脂乳液中残余单体含量的气相色谱测定方法探讨 吴亚虎,韩婷婷(广东中山市巴德士化工有限公司品控中心,528427) 摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 关键词:气相色谱;极性小口径毛细管柱;内标法; 醋酸乙烯酯、甲基丙烯酸甲酯、苯乙烯、丙烯酸丁酯、丙烯酸异辛酯等单体。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.实验部分: 1.1 仪器和试剂 电子分析天平(万分之一);GC5890F气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mm x 30m x 0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4+1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2 测定原理试样中加适量内标物并用少许丙酮(4+1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3 测定条件 气化温度:280℃ 检测温度:320℃ 载气:氮气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为60Kpa(30℃); 氢气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为65Kpa(30℃); 空气:变色硅胶[/fon
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=39639]GB/T 5009.204-2005 食品中丙烯酰胺含量的测定方法[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱(GC-MS)法[/url]
摘要:建立了毛细管气相色谱法测定酱油中甜蜜素含量的分析方法。采用RTX-WAX毛细管色谱柱和氢火焰检测器测定,外标法定量。样品加标平均回收率为93.7%-99.3%,相对标准偏差(RSD)为2.5%-4.7%。实验结果表明,该方法简便、快速,适合酱油甜蜜素含量的测定。关键词:酱油 甜蜜素 气相色谱甜蜜素又名环己基氨基磺酸钠(C6H12NaO3S),是用来增加食品甜度的化学调味品。由于甜蜜素的甜度只有糖精的三十分之一、口感好、价格低廉,被用来代替食糖在饮料、糕点、蜜饯等多种食品的生产中广泛应用。由于过量的甜蜜素等甜味剂会对人体健康产生一定的影响,如:损害人体的肝脏和神经系统等,因此,强制性国家标准GB2760-2007中明确规定甜味剂是限范围、限量使用的食品添加剂,其本身产品质量也有国家标准(GB12488-2008)。按照国家的有关规定,酱油中甜蜜素不得检出。 照国标《食品中环己基氨基磺酸钠的测定》(GB/T5009.97-2003)中规定的方法,目前对酱油采用液体试样的前处理方式,即称取样20g左右于100ml比色管,置冰浴中。加入5ml50g/L亚硝酸钠溶液,5ml100g/L硫酸溶液,摇匀,在冰浴中放置30min,并经常摇动,然后加入10ml正已烷,5g氯化钠,摇匀,待分层后吸出已烷层,离心后进行GC分析。但是,在实际操作过程中,以上样品存在着乳化现象严重、不易分离等缺点,影响了测定甜蜜素含量的准确性。因此,本文对酱油的前处理方法进行改进,探讨了采用固体试样的前处理方法及对气相色谱分析的影响,提高甜蜜素定量分析的准确性。1材料与方法:1.1仪器和试剂 岛津GC2010带FID检测器,配备A oc-20i 自动进样器和GCsoluthion 工作站, 长沙平凡仪器仪表有限公司:TG16-Ⅱ型高速离心机。正己烷(分析纯)、氯化钠(分析纯)、亚硝酸钠(分析纯),浓硫酸(分析纯)、RO水。标准溶液配制:甜蜜素标准品(中国计量科学研究院)用RO水配制成10mg/ml标准储备液。1.2色谱分析条件:气相色谱柱采用岛津公司RTX-WAX毛细管色谱柱,30m×0.25um×0.25rm(膜厚),分流/不分流进样口,进样温度200℃,分流进样,分流比10:1 载气:高纯氮气(纯度99.999%),氮气流速:40ml/min,空气流速:300ml/min, 氢气流速:30ml/min,进样量:2.0ul[
气相色谱仪如何测定苯乙酮的含量,我没学过很想知道,我们厂生产苯乙酮但是不知道怎么用色谱仪做,测定含量







 我要推广仪器
我要推广仪器
 下载APP
下载APP