各位专家: 我公司有一台GC920色谱仪,本来接的是软管,现在都更换了不锈钢管,接好后试了一下,结果主峰峰面积变得很小,跟杂质峰差不多,但前一天用得还好好的,一切正常,能检查的都检查了(气路正常,点火也正常,也没有漏气),但还量查不出什么原因,请各位专家指教,谢谢!
测试了ECD和FID检测器,在使用自动进样和顶空自动进样的时候,自动进样的状态下峰面积变小很多,顶空进样正常。怀疑是进样口出了问题,跟换了衬管,隔垫,密封圈,峰面积还是很小,换了新的色谱柱也还是同样的问题。现在真是心力交瘁??,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]是安捷伦7890B
[color=#444444]用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测同一种物质,进样量和温度条件都是相同的,但是为什么测出来的峰面积相差特别大??p.s.我是手动进样,跟这个有关系吗??[/color]
各位大侠好! 请不惜赐教一二!现用的是岛津GC-2010的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],柱子是RTX-1,L 30、ID 25、DF 25.本仪器专门用来做定量的,现有问题如下: 1.周一做定量预分析,新配置的0.075mg/mL 的标准溶液峰面积约为55956.3,分析样品溶液浓度约为71%,计算定量应称取质量为m(mg) 2.次日同一样品平行称取六份配置样品溶液浓度,与0.075mg/mL的标准溶液一同上机测试,结果标准溶液峰面积变为46655.6,样品浓度最终计算结果变为75.7%。但是出峰时间没变,三次测试平行性很好,只是峰面积降低了。 3.接着新配制0.1mg/mL和0.075mg/ml的标准溶液,结果显示0.1的峰面积约为46230.1,0.075的将为39896.5左右。以前做0.1的峰面积都为62394.9左右。 4.已于昨天将柱子老化,问题依然存在。 新手求各位大侠指点
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]为国产GC9790,色谱柱为se-54毛细管柱。[/color][color=#444444]有次拆下毛细管柱,再装上去之后就出现问题了,峰面积越来越小,同样溶剂,同样进样量,同一根针,同一个人操作。第一次进样,峰高正常为1200,过一个小时后再进第二针,峰高为800,峰面积降了;第三针峰高就600了。进样垫为新换的,装色谱柱的石墨垫圈为新换,拆过进样器的玻璃衬管也是好的,没有堵。色谱柱装上后也检漏,并且柱子是通的,不堵。请教工程师,说没遇到过这个问题。我该怎么办?[/color]
求助!!质量比与气相中的面积比是否有相关性??有谁做过这类情况的验证??分子量为100的药物,与分子量800的药物,按照质量比1:1的配比,在气相色谱中是否也体现出1:1的面积比列???
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]软件中有个校正类型里面有个评估类型,有峰面积,峰高,相对峰面积等等,其中还有个CE峰面积,请问CE峰面积代表的是啥
各位大侠,你们好!我初次使用气象色谱,测定天然气中各组分的摩尔比,现有如下问题,希望可以得到高人指点!1、色谱图中自动积分给出的峰面积比是指组分的摩尔比还是质量比,这是困扰我的一个最大问题!2、由于N2与CH4的峰距离很近,曾尝试降低柱箱温度,但是效果并不明显,请问有没有什么更好的方法??大侠们,请多多指教,不胜感激!!
气相色谱分析煤气成分,各种分析条件没有做更改,突然有一天所有成分的峰面积都扩大了三倍左右(做了将近10个样品),第二天又恢复到原来的峰面积,过了将近半个月的今天又出现了所有峰面积都扩大将近三倍左右的情况(分析第一个样品的时候峰面积还没有变化,从第二个样品开始都扩大了,扩大后的几个标样中的峰面积平行效果还挺好),请问谁知道可能原因。
[table=100%][tr][td]DB-5 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱原来测苯胺类物质1mg/l峰面积在50000左右,现在测多环芳香烃1g/L峰面积为500000左右,浓度增大了1000倍峰面积只增加了10倍,怎么回事?[/td][/tr][/table]
气相色谱峰面积重现性差什么原因?
各位大神,请教一下GC-FID色谱峰面积突然变小,可能是什么原因。具体情况:甲烷411ppm峰面积在30000左右,丙烷443ppm峰面积85000左右。但由于更换助燃气导致短暂熄火,开始使用的是压缩机压缩空气,未加纯化器,后换为除烃压缩空气。3min我左右,点火,通标气测试,发现相同浓度只有27000和80000。不知道是什么原因。一直用质量流量计给色谱进样。色谱柱不知是否有影响,色谱使用过程中色谱柱性能下降?检测器未拆下来清洗,不知是否有影响?请大神帮忙分析一下。
请教大家一个问题:我刚刚开始做气相色谱,用的是岛津2010,没有自动进样器,最近做样发现峰面积不是很稳定,我也很注意手动进样手法还有进样量(1微升),前天做面积99000,今天做92000,不知道怎么搞的?而且我发现基线今天做不稳定,虽然有程序升温(前天也是程序升温,基线也很稳)。是不是基线不稳自动积分也会导致面积有差距?说的有点乱,大家勉强得看吧,谢谢各位老师了。
气相色谱的峰面积都很大,比如谱图上显示某成分的峰面积是10268.241,这个数值应该是几位有效数字?肯定不是8位有效数字,因为这个数值是电脑算出来的,并不是根据有效数字运算规则算出来的。按气相色谱对峰面积的计算方法,这个数值应该保留几位?
各位版友,我所使用的仪器配置为:7694E的顶空进样器、7820气相、以及EZchrom的软件,在顶空进样时,溶液里面的氯苯与乙醇峰面积相对以前的标样来说小了几十倍,但是溶剂DMSO的峰面积变化不大,减小顶空进样器上面的分流阀后进样,DMSO峰变大,但是氯苯跟乙醇几乎无变化,不知道这两个物质被谁偷走了,在采用GC直接进样时峰面积跟以前变化不大!请问有哪位版友遇到该类型问题了?
关于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],请问各位大神,在色谱测量条件都不改变的条件下,峰面积和进样气体的温度有关系吗?我想在常温下测量标气的峰 反应气的温度达到了几百度,不知道误差会不会很大
请教大家一个问题:我刚刚开始做气相色谱,用的是岛津2010,没有自动进样器,最近做样发现峰面积不是很稳定,我也很注意手动进样手法还有进样量(1微升),前天做面积99000,今天做92000,不知道怎么搞的?而且我发现基线今天做不稳定,虽然有程序升温(前天也是程序升温,基线也很稳)。是不是基线不稳自动积分也会导致面积有差距?说的有点乱,大家勉强得看吧,谢谢各位老师了。
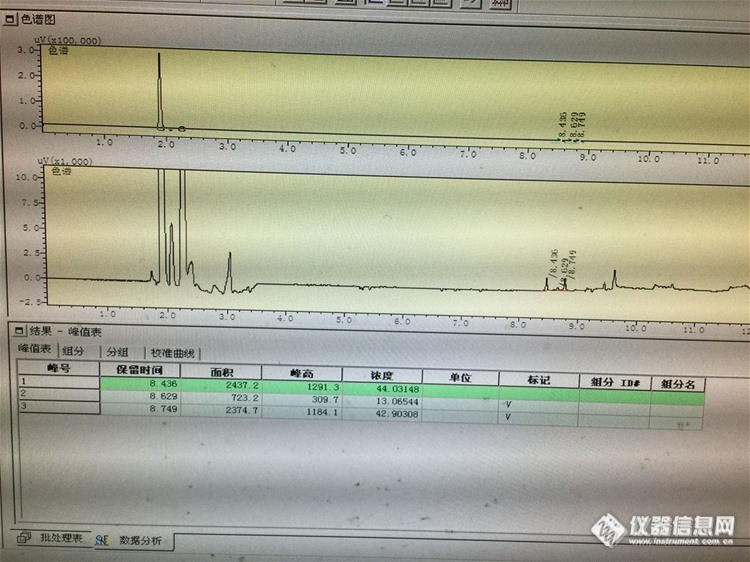
请问气相色谱仪,检测一个样品三次,有机氯检测,三次的峰面积偏差多少是正常的?软件积分一般都准确么?为什么会偏差比较大的数据都有的,是否可以手动积分?
[color=#444444]用安捷伦7890B检测液体样品,进样量1微升,分流比是20:1,采用自动进样方式,之前在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中打出的内标物峰面积都是1000左右,今天用同样的方法进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析,内标物峰面积变化很大,做了好几次,峰面积为50-60左右,偶尔有几次出现180+和200+,但都与之前的峰面积存在较大的差异,而使用手动进样进气体样品分析则不存在差异,请问是什么问题?如何解决?[/color]
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]ECD检测器,峰面积单位Hz.s,做期间核查时要换算成Hz.min,应如何换算

北分瑞利 SP-3420A气相色谱,最近做样品峰面积变小,一切检测条件不变的情况下,苯系物现在做浓度70的,跟前段时间浓度10的峰面积差不多,非甲烷总烃进标气,以前总烃峰面积五万多,现在五百多。。。这是什么原因呢?换了色谱柱也一样,检测器喷嘴擦了也没有改善。。。附几张对比图,一切检测条件都没变。。。http://ng1.17img.cn/bbsfiles/images/2016/09/201609011123_607816_3079083_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609011123_607817_3079083_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609011123_607819_3079083_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609011123_607818_3079083_3.png
急急急,请教各位老师,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做苯系物,分流比为20时,各个物质出峰峰面积特别小,0.25ppm的检测不到峰,但是溶剂峰特别大,峰面积有一百三十多万。如果继续改分流比,会导致溶剂峰更大,会不会对衬管进样口有影响呢?
请教各位大神 关于影响[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]峰面积RSD的因素的问题 顶空手动进样 做系统适用性 走对照溶液的时候 各组分峰面积RSD总是偏高 特别是刚开始进的第一针峰面积偏低很多
使用的安捷伦8860[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url] 更换了一个色谱柱后 分析氯乙烯中的乙炔和氯化氢含量 氯化氢数据还好 识别峰面积在2500左右 乙炔气几乎没有峰面积识别不出来是怎么回事 求助如何解决
同种物质在相同的色谱柱上,只通过变换温度,使这种物质的保留时间提前,会使这个物质的峰面积变大吗?(包括所有检测器)原因是什么,质量型浓度型有区别吗
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进样得到甲苯峰面积2万左右,如何计算此时甲苯的浓度呢?我是打一针1微升甲苯溶液,然后根据两者峰面积换算的,这样计算是不是有问题?[/color]
比如:谱图里给出一个组分的峰面积是11326.218,因为这个数值是电脑算出来的,并不是根据有效数字运算规则算出来的,那么这个数值应该是几位有效数字?肯定不是8位有效数字。谁知道气相色谱峰面积的计算方法?似乎只有根据这个计算方法,才能确定11326.218的有效位数吧。请高手指点一下,这个数到底该保留几位有效数字,11326.218中,从哪位数开始,它的数值已经是不确定数字(估计值),应该舍去。
[color=#444444]有样品1,里面含有物质A和B,然后换了不同的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱近10根,其他条件都是一样,样品一直都是同一个样品,为何在不同的柱子上,A和B的相对峰面积比值变化特别大,这是为何?比如HP-5柱上,A/B=1:2,然后SE-30上,A/B=3:2。[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]上确定,出峰顺序没有变化。求各位帮忙指导解惑?[/color]
[align=center][size=24px][b][url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]出现峰面积变大或平头峰等该如何解决[/b][/size][/align][align=left][size=18px] 理想情况下,经[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分离的峰应该为高斯分布曲线,即对称峰。但实际上当一个样品谱带沿着色谱柱前进时,由于浓度差等原因,样品分子会向谱带两侧扩散,从而使色谱柱出口处的样品谱带比柱入口处宽,且可能产生不对称的峰,就是谱带展宽。 一、不对称峰-谱带展宽 理想情况下,经[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分离的峰应该为高斯分布曲线,即对称峰。但实际上当一个样品谱带沿着色谱柱前进时,由于浓度差等原因,样品分子会向谱带两侧扩散,从而使色谱柱出口处的样品谱带比柱入口处宽,且可能产生不对称的峰,就是谱带展宽。谱带展宽的程度主要用柱效来表示,色谱峰越对称,峰越窄,柱效越高。 影响谱带展宽的因素有多种,主要分为柱内和柱外两种。柱内因素是指色谱柱本身的性能,如柱活性大小、固定相是否与样品发生化学反应、柱效是否够高、样品是否超载等。柱内因素导致谱带展宽的因素则主要是指街头的死体积、进样口和检测器死体积等。在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]中,柱外因素导致谱带展宽的程度要比柱内因素小得多。 二、峰面积变大 导致色谱峰峰面积变大的主要原因有以下几点: ①方法参数设置: a.分析条件的改变,如环境[url=http://www.ehsy.com/category-15806]温度[/url]升高,分流比变化等。分流比变小,峰面积变大。 b.数据处理机问题。比如重新开机后,可能会出现这种情况; c.方法设置参数变化,如积分参数变化; d.手动进样时进样技术不好。 ②仪器因素: a.载气流速控制不好,流速增大或柱前[url=http://www.ehsy.com/category-15808]压力[/url]调节阀异常,可能出现这种情况; b.分流口被污染; c.程序升温过程中升温重复性不好,柱温控制不良; ③色谱柱因素: a.色谱柱类型不适合分析该样品,导致固定液流失; b.柱温过高超过了色谱柱固定液的温度上限,导致固定液流失;柱温太靠近色谱固定液的温度下限,导致样品在流动相和固定相之间的分配比发生变化; c.色谱柱用了段时间,未老化,柱性能变差甚至有之前的样品残留物,可能导致这样情况; d.柱温未达到平衡就开始进样; 解决方案如下: 测定时色谱峰面积变大的现象,在确认没有改变色谱条件和方法参数的前提下,首先要考虑进样问题。如果是手动进样,要提高进样技术,进样量要准确、稳定。如果是自动进样,则维护、改善进样系统,保证进样器正常工作; 其次,考察仪器因素。要观察载气压力是否稳定、柱前压调节阀是否有问题;测定分流口和隔垫吹扫口排出的载气,排出气是否减少,必要时调整分流比或清洗分流口。 另外,要记录升温过程(柱温,如有程序升温的进样口也要考察)的温度变化,控温精度是否正常;柱温的控制是否正常尤其重要。 如有问题,需要维修温控系统的电路部分。当然,色谱柱的因素也必须考虑。色谱柱的固定液类型是否适合分离该样品,柱温是否合适等。如果色谱柱不合适,更换色谱柱。 如过色谱柱用了很久,没老化,就老化后再做;如果老化色谱柱,柱性能仍不能恢复,那么得更换新的色谱柱。 三、平头峰 色谱图中出现平头峰,首先要考虑进样量是否过大,导致信号过大,信号超过记录仪的zui大测量值,不再上升出现的平头峰,包括进样量过大及浓度太大等;还可能是检测器灵敏度选择太高,离子化检测器所用静电计输入达到饱和,记录仪滑线电阻或机械部分故障。 解决方案如下: ①减少进样量,或对样品进行合理稀释,或进样时加大分流比; ②适当调节检测器信号衰减,改变记录仪量程; ③增大色谱仪上衰减倍数,减小灵敏度。 当然,在色谱分析时,定量的依据是色谱峰响应大小与组分量在一定范围内呈线性关系。 对有些试验而言,主要考察的是杂质量,所以有时为了能准确检测出有关杂质化合物的量,往往会通过增大供试品溶液浓度,提高杂质的信号响应值来进行试验,这时供试品主峰就可能会出现平头峰,因杂质峰的响应值与主成分峰的响应值相差太大且无关,因此不必关注此类主成分平头峰,对杂质首选自身标准品对照定量法,实际试验中要注意具体问题具体分析。 四、圆头峰 色谱分析中出现圆头峰,有以下几个方面原因: ①进样量过大,超过检测器的线性范围(ECD检测时尤其如此); ②检测器受固定相流失及样品中高沸点成分、易分解组分及腐蚀性物质的污染; ③记录仪灵敏度过低; ④载气系统可能存在泄漏。 解决方案如下: ①减少样品溶液进样量或将样品稀释后再进样,或增大分流比来进样; ②清洗检测器,如果污染物仅限于高沸点物质,则通常可将检测器加热至zui高使用温度后,再通入载气就可清除,要注意加热的温度不能损坏检测器的绝缘材料; 如果加热法不适宜,也可以用丙酮等溶剂从进样口注入(每次可注入几十微升)进行清洗,在污染程度较轻时是有效的;若以上方法都不能解决污染问题,则应将检测器卸下,选择既能溶解污染物又不损坏检测器的溶剂,用注射器注入测量池进行彻底清洗; ③适当调节记录仪灵敏度; ④查看载气气路压力,仔细检查是否存在泄漏,这种情况一般伴随着保留时间或响应值的变化。[/size][/align]
在用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]定量分析时,为什么一般用峰面积定量?







 我要推广仪器
我要推广仪器
 下载APP
下载APP