[b][size=24px]气相色谱仪器图谱分析使用口诀[/size][size=18px]气相色谱分析法概述气液试样实在多,气相色谱把样测分析灵敏响应快,常量分析全包括内标外标归一法,检测热导氢焰化微机处理色谱图,定量结果准度大气相色谱仪气相色谱仪器多,型号性能各有别分析特性有异同,分离分析有特色载气系统稳操作,携带样气去检测压力流速勿波动,稳压稳流靠调节柱分系统分类多,填充毛细全包括分离试样全靠它,选择液载要正确进样系统严操作,定量进样需准确气体采用进样阀,液体微量用注射检测系统蕞明确,种类型号也很多热导氢焰离子化,电子捕俘双焰火检测信号需琢磨,各个组分细甄别响应快速能分辨,一一对应勿出错放大记录有把握,谱图精细准轮廓色谱峰大含量高,微机定量更准确利用气相色谱分析法定性和定量气相色谱分析法,试样定性较发杂操作相同峰一致,纯物对照判断它气质联用好办法,色谱红外能简化试样组分可判定,定性数据能表达气相色谱分析法,试样定量不发杂谱峰面积知多少,总量相比求得它定量测定常规法,内标外标归一化测定一种内外标,全部出峰归一化法归一化法来定量,出峰不全不适当全部组分看做一,百分之百来计量质量分数即含量,谱峰面积先测量校正因子查表得,代入公式得含量内标法定量可用内标法,分析简便不复杂出峰组分不完全,计算仍然要方法内标物质物含杂,称量不准出误差谱峰面积测准确,质量分数准度大外标法定量可用外标法,操作条件较复杂平行测定要保证,标准试剂用量大固定称量熟练化,称量不准出误差偶尔测定公式算,常规工作曲线查[/size][/b]
摘 要:为了找到焦炉煤气组分气相色谱法分析最优的分析条件,使用自装柱,通过正交实验设计,研究了分析 条件对分析结果的影响。结果表明:最优分析条件为:载气流速:43ML/min;柱箱温度:室温;检测器桥电流: 120mA;检测器温度:100℃。通过分析可得出如下结论:柱箱温度是影响分析的主要条件,而载气的流速、检 测器温度和检测器桥电流的影响并不显著。 关键词:组分分析;焦炉煤气;气相色谱法;装柱; 正交实验 0 引 言 焦炉煤气中含有多种组分,如甲烷、氢气、一氧化碳、氧气和氮气等。焦炉煤气中各组分含量关系到燃气的热量、华白数等一系列重要参数。因此,焦炉煤气中各组分含量的精确检测对于燃气生产和输配企业来说非常重要。气相色谱法作为一种高选择性、高效能和高灵敏度的分析手段,被广泛应用于各种气体的分析检测中。国家早在1989 年就制定了GB10410.1-89《人工煤气组分气相色谱分析法》国家标准。在几十年的应用中发现了不少问题,有很多作者对其进行了分析和改进,并与传统的化学分析法作了比较。但是,其中仍缺乏对分析条件系统研究,缺乏详细、系统的实验数据。国家在2009年又出台了新的国家标准GB/T 10410-2008《人工煤气和液化石油气常量组分气相色谱分析法》,并对相关内容进行了修改。在新出台的标准中柱箱温度的适用范围缩小了。这说明在旧标准所规定的温度条件值得商榷。在新标准出台之前,实验室的分析测试中也发现了同样的问题。另外,由于分析过程中, 焦炉煤气中CO2 在分子筛上存在不可逆吸附,分子筛遇水也会老化,因此,在实际测试过程中需要经常更换色谱柱。如果操作者在实验室能够自行填充色谱柱,则更为方便。针对以上问题,作者对色谱柱的填充过程进行了研究,自行填装了色谱柱。并使用自填柱,通过正交设计方法,讨论了分析条件对分析结果的影响,确定了最佳测试条件。 1 实验 1.1 实验仪器及试剂 气相色谱仪(;热导检测器(TCD)取样袋(光明化工研究设计院);标准气(北京兆格气体科技有限公司);氮气(鞍山鸿泰低温设备厂);氢气发生器(天津市分析仪器厂);样品取自鞍山市管道焦炉煤气。 色谱填料:13X 分子筛、GDX-104 填料(天津化学试剂二厂);空色谱柱(内径3 mm,长3 m 的色谱柱一根,装填13X 分子筛;内径2 mm,长2 m色谱柱一根,装填GDX-104 填料)(大连伟达分析仪器厂)。 标准气(? (CO2)=2.03%;? (CO)=7.12%;? (CH4)=30.4%;? (O2)=0.508%;? (N2)=9.19%;H2 为平衡气)(光明化工研究设计院)。 1.2 气相色谱柱的装填 首先用碱溶液将空柱管清洗干净,然后用清水将柱管中的碱液冲洗干净,放置到烘箱中烘干,待用。按一定的填充密度/ML), 根据柱体积计算所需的填料质量,并用电子天平称取,待用。 在柱的一端用玻璃丝绵堵住,用自制的装柱配件将柱连接到真空泵上,另一端通过装柱配件连接到柱头。将填料少量、多次地填到装柱漏斗中,并用真空抽吸,并不断震荡柱,使填料填充均匀。待柱装满后,将柱的另一端也用玻璃丝绵堵住,并标注填充方向。 在通氮气的条件下,将柱在200 ℃下,老化4 h,然后测试柱效和分离效果。 1.3 气相色谱法分析焦炉煤气成分条件的选择 由于焦炉煤气中含氢气、甲烷、氧气、氮气、 一氧化碳、乙稀和乙烷等多种气体,不能在一个分析条件下进行全分析。因此,需要在不同条件下对不同组分进行分析。本论文采用表1 所示的条件对焦炉煤气进行分析,其它分析条件则通过实验作进
仪器装置编辑气流系统 指载气及其他气体(燃烧气、助燃气)流动的管路和控制、测量元件。所用的气体从高压气瓶或气体发生器逸出后,通过减压和气体净化干燥管,用稳压阀、稳流阀控制到所需的流量。 分离系统 由进样室与色谱柱组成。进样室有气体进样阀、液体进样室、热裂解进样室等多种型式。色谱柱通常为内径2~3毫米、长1~3米、内盛固定相的填充柱,或内径0.25毫米、长20米以上、内涂固定液的开管柱。样品从进样室被载气携带通过色谱柱,样品中的组分在色谱柱内被分离而先后流出,进入检测器。 检测系统 包括检测器、微电流放大器、记录器。检测器(表3)将色谱柱流出的组分,依浓度的变化转化为电信号,经微电流放大器后,把放大后的电信号分别送到记录器和数据处理装置,由记录器绘出色谱流出曲线。 数据处理系统 简单的数据处理部件是积分仪。新型的气相色谱仪都有微处理机作数据处理。 温度控制系统及其他辅助部件 温度控制器用于控制进样室、色谱柱、检测器的温度。如果色谱柱放置在有鼓风的色谱炉内,则要求色谱炉能在恒定温度或程序升温下操作。重要的辅助部件有顶空取样器、流程切换装置等。 流动相 即载气 可用氦气、二氧化碳、氢气、氮气等。载气的选择与纯化的要求取决于所用的色谱柱、检测器和分析项目的要求,如对有些固定相不能与微量氧气接触,又如对热传导池检测器宜用 气相色谱法氢气作载气;对电子捕获检测器须除去载气中负电性较强的杂质,以利于提高检测器的灵敏度。用分子量小的气体作载气时可用较高的线速,这时柱效下降不大,却可以缩短分析时间,因为分子量小的气体粘度小,柱压增加不大,并且在高线速时可减小气相传质阻力。用氢气作载气时,在填充柱和开管柱中的流速可分别选用35和2毫升/分左右。 固定相 一般来说,宜按“相似性”原则选择固定液;分析非极性样品时用非极性固定液;分析强极性样品时用极性强的固定液(表4)。把固定液涂敷于开管柱的内壁,或涂渍在载体上制成填充柱的固定相,均勿太厚。开管柱的df宜为0.2~0.4微米,填充柱的固定液含量宜为3%~10%。载体颗粒约为柱径的0.1,即80~100目较好。这样,组分在液相中传质快载体粒度较小而又未增大填充不均匀性,有利于在较低的温度下分析高沸点组分及缩短分析时间。 操作温度 进样室的温度应根据进样方法和样品而定。气化方式进样时,气化温度既要使组分能充分气化,又不会分解(裂解进样除外)。检测室的温度以稍高于柱温为好,可避免组分冷凝或产生其他问题。色谱柱温的确定要作综合考虑,即要照顾到固定相的使用温度范围、分析时间长短、便于定性和定量测定等因素。最好能在恒温下操作,沸程很宽的样品才采用程序升温操作。满意的操作温度须由实验求得。 样品预处理 欲分析的化合物常用化学反应的方法转变成另一种化合物,这称为衍生物的制备。然后再对衍生物进行色谱分析。预处理的好处是:①许多化合物挥发性过低或过高,极性很小或热稳定性差,不能或不适于直接取样注入色谱分析仪进行分析,其衍生物则可以很方便地进入色谱仪;②一些难于分离的组分,转化成衍生物就便于分离和进行定性分析;③用选择性检测器检测可获得高灵敏度的衍生物;④样品中有些杂质因不能成为衍生物而被除去。 气相色谱法最常用的化学衍生物法有硅烷化反应法、酰化反应法和酯化反应法(有重氮甲烷法、三氟化硼催化法和季硼盐分解法等)。在制备化学衍生物时要特别仔细,否则会带来严重的错误。 内标准法 取标准被测成分,按依次增加或减少的已知阶段量,各自分别加入各单体所规定的定量内标准物质中,调制标准溶液。分别取此标准液的一定量注入色谱柱,根据色谱图取标准被测成分的峰面积和峰高和内标物质的峰面积和峰高的比例为纵坐标,取标准被测成分量和内标物质量之比,或标准被测成分量为横坐标,制成标准曲线。 然后按单体中所规定的方法调制试样液。在调制试样液时,预先加入与调制标准液时等量的内标物质。然后按制作标准曲线时的同样条件下得出的色谱,求出被测成分的峰面积或峰高和内标物质的峰积或峰高之比,再按标准曲线求出被测成分的含量。 所用的内标物质,应采用其峰面积的位置与被测成分的峰的位置尽可能接近并与被测成分以外的峰位置完全分离的稳定的物质。 绝对标准曲线法 取标准被测成分 按依次增加或减少阶段法,各自调制成标准液,注入一定量后,按色谱图取标准被测成分的峰面积或峰高为纵坐标,而以标准被测成分的含量为横坐标,制成标准曲线。然后按单体中所规定的方法制备试样液。取试样液按制标准曲线时相同的条件作出色谱,求出被测成分的峰面积和峰高,再按标准曲线求出被测成分的含量。 峰面积百分率法 以色谱中所得各种成分的峰面积的总和为100,按各成分的峰面积总和之比,求出各成分的组成比率。 10色谱分析编辑综述 从色谱图可以看到,色谱峰是组分在色谱柱运行的结果,它是判断组分是什么物质及其含量的依据,色谱法就是依据色谱峰的移动速度和大小来取得组分的定性和定量分析结果的。 定性分析 在给定的条件下,表示组分在色谱柱内移动速度的调整保留时间是判断组分是什么物质的指标,即某组分在给定条件下的t恼值必定是某一数值(图 1)。为了尽量免除载气流速、柱长、固定液用量等操作条件的改变对使用t恼值作定性分析指标时产生的不方便,可进一步用组分相对保留值α或组分的保留指数来进行定性分析。计算组分 i在给定的柱温和固定相时的保留指数Ii的公式为(公式4) 公式4式中n与n+1是紧靠在组分i前后流出的正构烷烃的碳原子数气相色谱法 是这两个正构烷烃的调整保留时间。 将样品进行色谱分析后,按同样的实验条件用纯物质作实验,或者查阅文献,把两者所得的定性指标(α值、t恼值或I值)相比较如果样品和纯物质都有定性指标数值一致的色谱峰,则此样品中有此物质。 由于只能说相同物质具有相同保留值的色谱峰,而不能说相同保留值的色谱峰都是一种物质,所以为了更好地对色谱峰进行定性分析,还常采用其他手段来直接定性,例如采用气相色谱和质谱或光谱联用,使用选择性的色谱检测器,用化学试剂检测和利用化学反应等。 定量分析 色谱峰的大小由峰的高度或峰的面积确定。可用手工的方法测量峰高,和以峰高h与峰高一半处的峰宽ω┩的乘积表示峰面积。A=hω┩。新型的色谱仪都有积分仪或微处理机给出更精确的色谱峰高或面积。应该注意,组分进入检测器产生的相应的色谱信号大小(峰高或峰面积)随所用检测器类别和载气的不同而异,有时甚至受到物质浓度和仪器结构的影响。所以须将所得的色谱信号予以校正,才能与组分的量一致,即需要用下式校正组分的重量: W=f′A式中f′为该组分的定量校正因子。依上式从色谱峰面积(或峰高)可得到相应组分的重量,进一步用下述方法之一计算出组分i在样品中的含量Wi:①归一化法将组分的色谱峰面积乘以各自的定量校正因子,然后按下式计算(公式5) 公式5此法的优点是方法简便,进样量与载气流速的影响不大;缺点是样品中的组分必须在色谱图中都能给出各自的峰面积,还必须知道各组分的校正因子。 ② 内标法,向样品中加入被称为内标物的某物质后,进行色谱分析,然后用它对组分进行定量分析。例如称取样品Wm克,将内标物Wφ克加入其中,进行色谱分析后,得到欲测定的组分与内标物的色谱峰面积分别为Ai和Aφ,则可导出:(公式6) 公式6此方法没有归一化法的缺点,不足之处是要求准确称取样品和内标物的重量,选择合适的内标物。 ③ 外标法在进样量、色谱仪器和操作等分析条件严格固定不变的情况下,先用组分含量不同的纯样等量进样,进行色谱分析,求得含量与色谱峰面积的关系用下式进行计算:(公式7) 公式7式中k媴是组分 i单位峰面积百分含量校正值。此法适用于工厂控制分析,特别是气体分析;缺点是难以做到进样量固定和操作条件稳定。 11分析方法编辑分析方法实际上是在某一特定的气相色谱分析中使用的一系列条件。建立分析方法实际上是确定对于某一分析的最佳条件的过程。 为了满足某一特定的分析的要求,可以改变的条件包括进样口温度,检测器温度,色谱柱温度及其控温程序,载气种类及载气流速,固定相,柱径,柱长,进样口类型及进样口流速,样品量,进样方式等。检测器还可能有其它可供调节的参数,这取决于所使用的检测器类型。有一些气相色谱仪还有可以控制样品与载气流向的阀门,这些阀门开启与关闭的时间也可能对分析的效果有重要影响。 右图为GeoStrata Technologies生产的Eclipse气相色谱仪。它以三分钟为周期持续运转。该仪器有两个阀门,用来控制载气进入定量管。当定量管充满样品气后,切换阀门,载气就会通过定量管。载气的压强会将样品带入到色谱柱中进行分离。 载气选择与载气流速 典型的载气包括氦气、氮气、氩气、氢气和空气。通常,选用何种载气取决于检测器的类型。例如,放电离子化检测器(DID)需要氦气作为载气。不过,当对气体样品进行分析的时候,载气有时是根据样品的母体选择的,例如,当对氩气中的混合物进行分析时,最好用氩气作载气,因为这样做可以避免色谱图中出现氩的峰。安全性与可获得性也会影响载气的选择,比如说,氢气可燃,而高纯度的氦气某些地区难以获得。(参见:氦气——分布与生产) 很多时候,检测器不仅仅决定了载气的种类,还决定了载气的纯度(虽然对灵敏度的要求也在很大程度
各位大虾们,请教一下,气相色谱用外标法做实验分析的时候为什么引进工作曲线?引进后如何通过工作曲线来测出试样中未知物的含量?感谢大姐、大妹们的帮助,谢谢。
1主题内容与适用范围在石油加工和自然气净化工艺中产生大量的硫化氢气体,产业上普遍采用克劳斯法处理含有硫化氢的酸性气体,这种方法只有当H2S:SO2为2:1时,才能获得最高转化率。要获得较高的转化率,必须对H2S和SO2进行分析,并且要求分析结果正确可靠。此外,在克劳斯反应过程中还有一些副产物天生,如COS、CS2以及由空气带进的 O2、N2等。为了正确的把握这些组分的含量,对其比率进行最优化控制,减少副反应,进步硫回收率,确定了硫回收过程组成分析的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法。该法已用于硫磺回收过程气中的H2S、SO2、COS、CS2、O2、N2等组分的分析。2仪器与材料[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]。色谱仪应具有下列特性参数。检测器:热导检测器 气化室:要求在本方法使用的温度下能连续运转,为了减少其体积,它和色谱仪的连接间隔应尽量接近 记录仪:积分仪或色谱数据工作站 色谱柱:要匹配双柱,以补偿柱子流失产生的基线漂移 流量控制器:装备有稳流阀和稳压阀,先稳压后稳流 注射器:分格为0.01mL的1mL注射器和100mL注射器。2.2材料担体:分子筛和高分子多孔小球 载气:氢气(纯度≥99.99%) 标准气:H2S、SO2、COS、CS2、O2 稀释气:高纯氮(纯度≥99.99%)。3实验方法为了兼顾硫化物和氧气的分析,选择了如下实验条件。3.1色谱柱和固定相的选择不同的柱材料、柱型、柱长和柱效具有一定的影响。不锈钢柱的优点是不易变形而且具有一定的惰性,用它分析O2、N2、H2S、SO2、COS和CS2等一般化合物是足够稳定的。但是,若使用固定相Chromosorb101时不应使用不锈钢柱。为了进步柱效,色谱柱的外形采用螺旋管,柱内径为 2~3mm,螺旋管直径比色谱柱内径大15~25倍,柱长取2000~3000mm。3.1.2固定相的选择目前,气固色谱中使用的固定相主要有各种形式的碳分子筛和高分子小球等。分子筛是气固色谱中很重要的一种吸附剂,主要用于O2、N2、CO、CH4等永久气体的分离。在使用分子筛前要在真空下350℃左右干燥3h,以便除往水分和其他吸附物。本实验测得了不同温度下2种分子筛的吸水量,如图1所示。当分子中水分在9.37%时,氧和氮已不能完全分开,因此就要活化了,而活化的好坏会直接影响分离效果。分子筛的型号不同性能也不同,实验证实,在相同的条件下,5A分子筛和13X分子筛的保存性能不完全一样,在13X分子筛上 O2的保存时间较短一些,但是5A分子筛的分离能力却比13X分子筛强。高分子多孔小球可用作[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]的固定相,常用的牌号有 Porapak系列,Chrmosorb101、102系列,GDX第列等。GDX是有机芳香族碳氢化合同物,高温下使用体积会收缩,使填充柱产生裂层,在使用前活化会消除此现象,活化温度应高于使用温度20%左右。GDX301是中国自行研制的固定相,主要特点是疏水性强,耐腐蚀、热稳定性好,无固定相流失,可配合高灵敏度检测器使用。通过实验,对分析O2、N2、H2S、SO2、COS和CS2的固定相的种类、色谱柱的长度和材质条件进行了选择优化,结果表明:柱Ⅰ适应于分析H2S、SO2、COS、和CS2 柱Ⅱ适应于分析O2、和N2(见表1)。表1色谱柱及固定相的选定色谱柱柱长柱内径柱材质固定相分离组成编号/mm/mm──────────────────────────────Ⅰ20000.5不锈钢GDX-301H2S、SO2、COS、CS2Ⅱ30000.5不锈钢5A分子筛N2、O2──────────────────────────────3.2色谱柱和检测器温度的控制在50~100℃作了柱温选择实验。当柱温为50℃时,O2、N2分离良好,但H2S、SO2、COS、CS2峰形拖尾较明显,分析时间长,约 15min才能完成分析。当柱温为100℃时,硫化物分析时间可缩短为7min且峰形完整,但O2、N2分离不彻底。结果表明,柱温保持在80℃较适宜。检测器是一种指示并测定载气中被分离组分的装置。实验表明,检测器温度太高会引起噪音的增加,降低检测器的灵敏度,所以,检测器温度控制在100℃较为理想。3.3载气和流速的选择用H2作载气时柱效略低于用氮气作载气,但要缩短分析时间。载气流速对热导检测器的信号值有一定的影响,在热导检测上随载气流速的增加信号即有利于硫化物的分离,又兼顾到O2、N2的分离。3.4桥流在100~600℃mA范围内对桥流作了选择实验,增大桥路电流能明显地增大输出信号 然而桥路电流太大会使基线不稳定,噪音也相应增大。结果表明:采用140mA桥流可满足分析需要。3.5定性及定量分析3.1.5定性分析采用已知纯物质的标准气与未知样品的保存特性进行组分鉴定。即比较已知物和未知物的保存值(保存时间和保存体积),就能确定某一色谱峰代表什么组分。根据保存特性进行鉴定,是最常用的方法。3.5.2定量分析由于各种物质的峰面积与色谱条件有关,和物质的结构和性质没有固定数目关系,因此在进行定量分析时必须要用标准物质进行对比。采用工作曲线定量法或外标法定量。3.6外标气的配制在一个100mL医用注射器内加进少许硬质聚四氟乙烯块,注射器口用一软质橡皮帽封住,先吸取60mL高纯氮,然后用10mL注射器注进一定量的标准气,最后用高纯氮烯释到满刻度,反复摇动,负气体混合均匀。3.7校正曲线的绘制定量进样校正曲线是色谱分析常用的一种简便、快速的尽对定量方法。将已配制好的不同含量标准样,注进[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],然后将分析结果绘制成峰面积-百分含量校正曲线。在作硫磺回收过程气分析时打进同样体积的分析样品,根据所测得的被分析组分的峰面积值,可直接从外标校正曲线上查出相应组分的百分含量。4灵敏度、误差及最低检测浓度4.1灵敏度目前通常采用迪姆巴特(Dimbat)及其同事提出的方法来测定检测器的灵敏度,即表示为单位浓度的样品所导致检测器的响应值。操纵条件为桥流:140mA 柱温:80℃ 载气:氢气 衰减:1 检测器温度:100℃ 汽化室温度150℃ 载气流速:28mL/min 进样量:1mL。硫化氢为样品,测得:灵敏度S1900mV.mL/mL。4.2正确度表2列出了实验所测得的正确度的具体数据。4.3最低检测浓度在实验条件下实测最低检测浓度见表35结论(1)硫化物分析可采用气固和气液色谱法,但在工艺控制分析中采用气固色谱法具有无固定相流失、柱寿命长等特点,同时,也不会因柱室超温而损坏柱子,可用于高灵敏度检测器。实验证实GDX-301是分析硫化物的理想柱子 (2)由于硫化氢、二氧化硫、二硫化碳和硫氧碳都是极性物质,在分析过程中轻易发生吸附现象而影响分析结果。因此,应尽量缩短采样时间,确保分析结果的正确性 3在分析低含量氧时,应采取有效的密封措施,防止空气中氧的渗透。该方法是为硫磺回收过程气的分析而开发的,方法快速、正确、灵敏,分析误差为±1.4%,试验证实满足硫回收过程气的工艺分析。
RCONH22 确定初始操作条件主要包括进样口温度、检测器温度、色谱柱温度和载气流速。分流进样的进样量一般不超过2μL ,最好控制在 0 .5 μL 以下 ,进样量还和分流比有关 ,分流比大时 ,进样量可大一些 ;进样口温度应接近或高于样品中最重组分的沸点 ;对于一个未知的新样品, 可将进样口温度设置为 300 ℃;常用毛细管GC 所用柱内载气线流速为:氮气 20~40 cm/s。隔垫吹扫设定为 2 ~5 mL/min , 分流比依据样品情况(如待测组分浓度等)、进样量大小和分析要求来改变, 选择一个合适的折衷分流比,用分流比范围 20∶1 ~200∶1 ,待测组分浓度大或进样量大时, 分流比可相应增大,反之则减小,用大口径柱时分流比小一些,用微型柱做快速GC 时,分流比要求很大,流比小时, 分流歧视效应可能小,但初始谱带(主要是溶剂谱带)宽度大,分流比大时,初始谱带(主要是溶剂谱带)宽度小,但分流歧视效应可能大。检测器温度可参照色谱柱的最高温度设定,而不必优化。色谱柱温度,组成简单的样品最好用恒温分析;组成复杂的样品,常需要用程序升温分离;色谱柱的初始温度应接近样品中最轻组分的沸点, 最终温度取决于最重组分沸点;升温速率依样品的复杂程度而定,建议毛细管柱的尝试温度条件设置为OV -1或SE-54 柱 :从 50 ~280 ℃,升温速率 10 ℃/min ,V - 17(OV -1701)柱:从60 ~260 ℃, 升温速率 8 ℃/ min ,PEG -20M 柱:从60 ~200 ℃,升温速率 8 ℃/ min 。这是方法开发时的初始参考条件,具体工作中再根据样品的实际分离情况来优化设定。3 尝试性分析上述初始条件设定后,便可以进行样品的尝试性分析。一般先分离标准样品,然后分析实际样品。在此过程中,还要根据分离情况不断进行优化。GC的分离优化就是要在保证分离度和灵敏度的前提下,实现快速分析。在实际工作中,一般是首先满足分离度的要求,然后提高分析灵敏度,最后再考虑尽可能缩短分析时间。改变柱温和载气流速可改变分离度;内径越小,或者填料粒度越小,柱效越高;薄液膜色谱柱的柱效高于厚液膜柱;更换色谱柱可改变分离度;用化学作用如通过生化反应改变待测物结构;程序升温是GC分离复杂混合物的有效方法;进样量小一些、进样口温度高一些、载器气流速快一些、汽化室体积小一些,分流比大一些,对窄的初始谱带宽度有利。4 气相色谱定性与定量分析对于简单的样品,可通过标准物质对照来定性。对于复杂的样品, 则要通过保留指数定性和或GC/MS来定性。对于基层监测站,气相色谱定性分析最主要是依据保留值定性,即在相同的条件下,分别注入标准样品和实际样品,根据保留值确定色谱图上哪个峰是要分析的组分。但必须注意,在同一根色谱柱上,不同的化合物可能有相同的保留值,对未知样品的定性仅仅用一个保留值还不够。双柱或多柱保留指数定性是气相色谱定性分析较为可靠的方法,不同的化合物在不同色谱柱上具有相同保留值的几率要小的多。建议对复杂的样品采用双柱或多柱保留指数法定性。气相色谱定量方法包括面积百分比法、归一化法、外标法、内标法、标准加入法。基层监测站最常用的方法是外标法,只要用一系列浓度的标准样品做出工作曲线, 就可以在完全一致的条件下对未知样品进行定量

气相色谱-质谱法分析康酿克油的芳香性成分摘要:采用水蒸气蒸馏法从葡萄酒压榨渣中蒸馏提取得到精油。其气味芳香宜人。我们首次利用气相色谱-质谱联用分析其芳香性成分,通过NIST谱库检索,结合自建谱库,鉴定了其中38个组分,采用峰面积归一化法确定了各组分的质量分数,占色谱总流出峰面积的96.1%。芳香性成分主要为酸类,酯类。占总成分的99%左右。本项研究为康酿克油的开发利用奠定了基础。关键词:康酿克油 气相色谱-质谱Abstract: By steam distillation from the Wine squeezing slag extractedessential oil distillation. The smell fragrant. We first analysis the aromaconstituents using gas chromatography-mass spectrometry(GC-MS).Basedon the spectra search function of GC-MS with the aid of gas chromatographicretention rules,38 constituents were identified successfully. The relativecontent of the compounds were determined with peak area normalization method.The identified compounds constitute more than 96.1% of the total ion current.Esters are major constituents in the essential oil, representing 99%of thetotal contents. The basis for further developing the Cognac oil had been provided. Keywords: Cognac Oil, GC-MS 葡萄酒是用新鲜的葡萄或葡萄汁经过发酵酿成的酒精饮料。通常分红葡萄酒和白葡萄酒,气泡酒,三种。前者是红葡萄带皮浸渍发酵而成;后者是葡萄汁发酵而成的. 法国最古老的超一级酒庄是吕萨吕斯酒堡。我国葡萄酒的酿造起步较晚,但是进步很快。天津王朝葡萄酿酒有限公司就是一家很具规模的中法合资公司。天然康酿克油为制造葡萄酒时,在酒泥或压榨渣中提取的一种副产物,经过精馏可以得到纯度很高的精油。本实验首次对天然康酿克油的芳香性成分进行系统分析,为康酿克油的开发利用奠定了基础。1 实验部分1.1 材料和方法取天津王朝葡萄酿酒有限公司生产基地酿酒后的葡萄压榨渣1000g,进行水蒸气蒸馏。所得油层和水层用乙醚萃取3次,合并乙醚萃取液,用无水硫酸钠进行干燥,挥发掉乙醚后得到黄绿色蜜甜香且有葡萄酒香气味的精油,得油率约为0.08%(w/w)。1.2 仪器和实验条件仪器:气相色谱-质谱联用仪,型号:6890-5975(美国安捷伦公司,带自动进样装置)色谱条件:HP-5毛细管柱,(60m*0.25um*0.25nm,Agilent公司)以氦气为载气,恒流流速1.0mL/min,进样口250℃,分流比100:1,进样量0.2uL,升温程序:50℃保持5分钟,以3℃/min升到180℃,然后以15℃/min升至260℃。最后280℃保持10分钟质谱条件:接口温度280℃,电离方式EI,电子轰击能量70eV,离子源温度230℃,四级杆温度150℃。溶剂延迟4min,全扫描范围33-330amu,NIST05谱图与自建谱库检索。2 结果与讨论2.1 分析结果按上述条件对康酿克油进行GC-MS分析,经计算机检索NIST05标准谱库与自建谱库,也结合人工解析和查对先关资料 ,鉴定了其中38个成分,用面积归一化法计算出各成分的相对含量,结果见表1.表1 康酿克油的化学成分和相对含量 序号 保留时间/min 成分名称 分子式 相对含量 % 1 5.050 3-甲基丁醇 Isoamyl alcohol C5H12O 0.384 2 5.587 丁酸 Butyric acid C4H8O2 0.010 3 5.816 丁酸乙酯 Butyric acid ethyl ester C6H12O2 0.241 4 6.084 乳酸乙酯 Ethyl lactate [align=
请问一下大家,我用顶空-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]归一化法定量分析乙酰丙酸脱羧后丁醇和丙醇的产率,得到最后产率数据,那怎么根据数据作图??
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析中鬼峰产生的原因及排除方法?
[color=#444444]本人在做职业卫生方法验证,在做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法试验中,发现低浓度样品代入标准曲线,得出结果与理论值相差几倍,如果标准曲线强制归零后,结果正常,请问各位高手,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法回归曲线可以强制归零吗?多谢了!(我们的仪器是岛津GC2030)[/color]
[font=Encryption][color=#898989]摘要:[/color][/font][font=Encryption][color=#666666] 原油中烃类[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的传统方法是先对原油进行族分离得到饱和烃和芳烃,然后分别进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析,因此不能反映原油的原始特性和烃类组分的全貌.全二维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法能直接对原油样品进行分析,同时获得饱和烃和芳烃的地球化学参数,避免原油样品经族分离带来的分析误差,缩短分析时间.通过一维、二维色谱柱及升温速率、柱流量、初始柱温等条件实验,优选出原油中烃类全二维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的最佳实验条件 应用原油的全二维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/飞行时间质谱分析结果,建立了饱和烃、芳烃、萘系列、菲系列及二苯并噻吩化合物的相对位置定性图版,再利用图版对原油中组分进行定性分析 采用面积归一化法进行定量分析,并计算出了石油天然气行业标准所规定的饱和烃与芳烃地球化学参数 精确度实验结果表明,质量分数大于1%的样品多次重复分析的相对偏差小于5%,符合原油实验测定要求.[/color][/font]
[b]一、固定相的优选[/b]随着大量的液体固定相(超过1000种)的应用,大多数科学家并不相信所有这些固定相都能提供独特的分离性能,是否需要那么多固定相变成了一个问题。从1956年伦敦[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]会以来,此问题一直是争议的主题。许多[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]是相似的,具有类似的麦氏常数,如在1969年发表的1472个[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分离,几乎80%在五种不同化学结构的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]上取得。太多的固定相限制了不同实验室之间的交流和再现。[b]二、固定相的选择1.利用罗尔施奈德(R)/MCReynolds(M)系统[/b]对于含不同官能团的化合物的分析,R/M系统是有帮助的例如当需要对醇的保留作用比对芳烃大的柱子时,应选用Y与X之比值较高的固定相;为了得到对醇的保留作用比酮大的柱子,须选用Y与Z之比值较高的相。又如,对醇/酸的分离,可通过比较Y值;而对酮、醚、醛、酯,可看Z值;硝基化合物和氰基化合物,看U值N杂环,看S值。不仅如此,根据R/M系统还能指出柱子的使用价值。如果OV-1和OV-17能给出类似结果,则麦氏常数介于两者之间的固定相如OV-1也能给出类似结果。如果试验过一种固定液但分离效果不好,则最好选 McReynolds常数之和差异在200以上的固定液。[b]2.利用相似相溶原理[/b]固定液的性质与被分离组分之间有某些相似性如官能团、化学键、极性、某些化学性质等。性质相似时,两种分子间的作用力(色散力、诱导力、定向力或氢键作用力等)就强被分离组分在固定液上的溶解度就大,分配系数就大,因而在柱内的保留就长。须知,对于高沸点或挥发性较小物质,因其流出困难,实际上不宜选择与样品很相似的固定相,否则将造成出峰时间过长、操作温度过高等一系列问题。[b]3.利用优选固定相[/b]现在已有上千种固定相,而且还在不断增加这虽然加宽了选择范围,增加了选择的灵活性。然而,在目前尚无类似“对号入座”这种简易选择方法的状况下,固定相的型号过于繁多,却给选择带来了巨大的工作量,因此不少色谱分析工作者致力于固定相的优选工作。 Leary等于1973年用最相邻技术优选了固定相,依据最相邻距离优选出十二种固定相(图1)。填充柱中,最受欢迎的为:①甲基硅氧烷;(OV-101、SE-30等);②50%苯基甲基硅氧烷(如OV-17)③Carbowax 20M④中等氰基含量的氰基相(OV-275、SP-2330、Silar5CP)⑤50%三氟丙基硅氧烷(如OV-202、SP-2401)对毛细管色谱,最受欢迎的为:①甲基硅氧烷② Carbowax20M③氰丙基硅氧烷或三氟丙基硅氧烷。[img=image.png,398,270]https://i3.antpedia.com/attachments/att/image/20220126/1643186364540316.png[/img]图1 与Squalane的最相邻距离括号中数字为优选相与Squalane的距离对无机气体和轻烃,高分子多孔微球如 Porapaks是优选相。我们曾经总结27条气体分析柱系统选择规则,引述如下:1)对一般永久性气体的分析建议采用5A Molsieve PLOT柱,这些气体包括He、Ne、Ar、Kr,Xe、Rn、H2、CO、NO、N2O、O2、N2、CH4、CD4、C2H6等,但样品中必须保证无H2O、CO2、SO2及H2S等重组分。2)如果要分离O2/N2,同时样品中又含H2O、CO2等重组分,建议采用 Carbo PLOT 007柱,C1~C3也可流出。3)含极性和非极性组分的混合物的分析,建议采用 Pora PLOT柱,此柱也适用于永久性气体分析。能在此柱上分析的组分有:水汽:CO、CO2、H2S、SF6、N2、O2、CH4、N2O、NO、Ar、NO2、SO2、NH3、Cos、 Freons、ethyl Oxide。烃:C1~C10。醇、酮、二醇、卤代烃以及通常在 Porapak Q、Haysep Q或 Chromosorb 102上分离的任何其他样品。不过,对几何异构体及0、m、p位置异构体及含活泼氢的组分,不如采用GLSC的石墨化炭黑WCOT柱。4)对化工原料中十分重要的正丁烯、异丁烯的分离,不如采用Al2O3/PLOT柱好。后者对C1~C9,轻烃中烯烃异构体的分离有高选择性。样品中不能存在水分、CO2。如果C10烃存在,就要用厚膜甲基硅氧烷交联柱,它对低碳饱和/不饱和烯烃异构体的分离,不如Al2O3/KCl好。5)对于不要求分析样品中所含少量CO2、H2等重组分,而要分别定量H2、O2、N2、CH4、CO的,如要快速分析,则用13X分子筛如要CH4、CO间有较大分辨率如痕量分析,则用5A Molsieve。6)如果采用低温,则H2、O2、Ar、CO也能用 Porapak分离,且在低温或长柱时,5A也能分析这些气体,不过出峰次序有所不同。因此,当分离O2中痕量Ar时,建议用5A Molsieve Porapak,当Ar/O2含量差不多时,两者均可,但用5A时O2/N2间分辨率大。7)当所用仪器无低温分离设备,而要求O2/N2分离,同时要求分离H2O、CO2或CH4C2H6等重组分,建议采用碳分子筛,等温和程升均可。8)如果设备可用低温,要分离O2/N2、O2/r的同时,还要分离N2O、NO、CO、CO2第四章[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]固定相H2S、COS、SO2等重组分,推荐 Porapak Q。9)如果有低温程升设备,也可采用 Porapak R,对烃的分离可一直进行到C9馏分:不过,对碳氢化合物的分离情况,当然不如用厚膜毛细管柱好。10)惰性气体(He、Ne、Ar、Kr、Xe等)的分离,最难分的是He、Ne,常温下用分子筛即可,其中,He、Ne、H2、O2,N2的分离用5A Molsieve好; He、Ne、H2,O2、N2、COCH4的分离用13X好。11)化工原料中十分重要的裂解气及液化石油气的分离,须用特殊选择性柱子。其中首推Picric acid改性的Carbopack柱,同时Durapak n-Octane,PorasilC或GDX-501等均能达到目的。12)为了分析天然气样品中含有的烷烃和直到C9、C10的重烃,采用非极性厚膜交联柱可取得良好的分离。13)如要进一步分离CO2和水分,也可采用和Porapaks 串联的切换性,如Porapak Q+ Porapak T/OV-1 或Porapak R/OV-101,14)当进一步要分离O2/N2时,可采用13X分子筛、 Porapaks及非极性毛细管柱组成的三柱切换系统。15)如果采用低温,甚至单根 Porapak R填充柱就能实现上述目的,对烃的分离当然不如非极性毛细管柱。16)合成气或可燃性气体的组成为H2、O2、N2、CO、CO2、CH4、C2H4、C2H6、C3+、2S等,因而有关这些气体的柱系统推荐规则,可根据分析目的选用规则1)~15)。17)对含氟的F3Cl、SF6等腐蚀性化合物的分析,由于腐蚀性强,只能采用合氟化合物为基质的担体和含氟化合物氟油为基质的固定液(氟油)来进行分析,所用柱管要用聚四氟乙烯(Teflon)。18)分离F2、O2、N2时,也可采用聚四氟乙烯柱系统,但应采用长柱,而F2O2分离,要采用银柱吸收的方法。19)聚四氟乙烯的柱型也可用于含氟的硫化物和金属含氟甚至含氟、含硫化合物的分析,其他腐蚀性的无机卤化物也要采用聚四氟乙烯柱系统。20)对于C1、C2有机气态卤化物的分离,如果用填充柱,建议采用 Chromosil 310。如果用毛细管柱,建议采用厚膜 Carbowax 20M交联柱。21)对于COS、H2S、CS2、SO2的分离,视H2S与CS2含量多少,可分别采用 Chromosil310、330或 Porapak QS Supelpak QS,其中前二者适于H2S含量高的情况,后二者适于CS2合量高或含水的情况。22)除上述气体外,采用 Chromosil 330或PoraPak QS也可分离C1~C3硫醇和硫醚,uL/L级也行。 Polyphenyl ether/H3PO4 on Chromosorb也可用于分离uL/L级以下的上述气体。当痕量分析时,柱管需用 Teflon。23)使用 Carbopack B/XE-60/H3PO4填料,在玻璃柱上即可实现对C2硫化物异构体的基线分离,费用比 Polyphenyl ether 低。这一柱系统也适用于SF6、H2S和COS的分离,这在Polyphenyl ether上是不可能的。24)对氮和它的氧化物N2O、NO的分离,建议采用5A Molsieve柱或 Porapak柱,但需注意NO2的反应性。25)碱性氮化物如氨、甲胺等的分离,建议采用 Chromosorb 103。26)对于同位素的分离,只要质量上有区别,就会使色散力有差别,因此可用高效[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱进行分离,这种柱子一般比较特殊,如:a、氢的同位素的分离,可采用Fe(OH)3腐蚀的Al2O3填充柱或5A Molsieve PLOT石英柱(75m×0.32mm),它们均可使H2、D2、HD2、HT、T2、DT获得分离。b.氮的同位素的分离,可采用60m×4mm的0.1% Squalane on Graphitised Carbon black柱,使14N、15N得到分离,载气H2中加进CO以失活表面。相同的填料也适用于-78℃时CH4与CH3D的分离。c.当采用NaOH腐蚀的47m×0.22mm玻璃毛细管柱,并用N2-He作载气,可分离甲烷的同位素化合物。64mx0.22mm的活性硅胶柱也行。d.氧的同位素的分离,由NaOH腐蚀的173m×0.3mm玻璃毛细管柱,并用N2-He作载气,可分离16O2和18O2。e.氖的同位素的分离,采用82m经腐蚀的玻璃毛细管柱在-254℃可分离20Ne和22Ne,载气为He+5%H2。由于[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]分离同位素不是最方便的办法,因此用得不广泛。27)硅烷、硼烷、镓烷及磷烷和砷化氢等,有很大毒性,但在色谱上的分离并不困难,多孔聚合物或非极性柱均可。硅烷中氯硅烷也能由非极性的 Squalane柱测定。[b]4.利用特殊选择性固定相[/b]特殊选择性固定相对特定类化合物具有特殊选择性。被分离组分与固定液分子之间往往有某些络合物或中间体生成,属于分子间化学作用的结果。常见的有:①硝酸银固定相保留烯烃;②重金属脂肪酸酯保留胺类;③有机皂土对芳烃位置异构体的选择性保留;④液晶对长/宽比不同的异构体的独特分离能力;⑤手性柱对旋光异构体的选择性分离等。尽管它们均有局限性,但从实用角度看,具有一定的价值。一些商品化的特定样品的专用柱或标准方法中限定的专用柱也可认为是属于此类固定相。关于[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱的应用和方法指南的更多信息可从大的色谱公司的产品目录看到,如安捷伦在其产品目录中给出了EPA方法中的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱、美国药典(USP)[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]固定相、ASTM方法[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱等
[url=http://www.gdkjfw.com/][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url][/url]内标法: 在分析测定样品中某组分含量时,将一定重量的纯物质作为内标物加到一定量的被分析样品混合物中,然后对含有内标物的样品进行色谱分析,分别测定内标物和待测组分的峰面积(或峰高)及相对校正因子,按公式即可求出被测组分在样品中的百分含量。加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。[align=center][img]http://www.gdkjfw.com/images/image/55461526982266.jpg[/img][/align] 内标法在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]定量分析中是一种重要的技术,是色谱分析中一种比较准确的定量方法,尤其在没有标准物对照时,此方法更显其优越性。 使用内标法时,在样品中加入一定量的标准物质,它可被色谱柱所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。 [url=http://www.gdkjfw.com/][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url][/url]外标法: 用待测组分的纯品作对照物质,以对照物质和样品中待测组分的响应信号相比较进行定量的方法称为外标法。例如,在色谱法中,想知道被测样品浓度。可以用外标法首先用待测组份的标准样品绘制工作曲线,测出各峰的峰高或峰面积对应的样品浓度,绘制出标准曲线。实际应用时,测出峰高或峰面积对应标准曲线,就可以得到样品浓度。[align=center][img=,627,382]http://www.gdkjfw.com/images/image/25681526982266.jpg[/img][/align] 此法可分为工作曲线法及外标一点法等。 工作曲线法是用对照物质配制一系列浓度的对照品溶液确定工作曲线,求出斜率、截距。在完全相同的条件下,准确进样与对照品溶液相同体积的样品溶液,根据待测组分的信号,从标准曲线上查出其浓度,或用回归方程计算,工作曲线法也可以用外标二点法代替。通常截距应为零,若不等于零说明存在系统误差。工作曲线的截距为零时,可用外标一点法(直接比较法)定量。 外标法方法简便,不需用校正因子,不论样品中其他组分是否出峰,均可对待测组分定量。但此法的准确性受进样重复性和实验条件稳定性的影响。此外,为了降低外标一点法的实验误差,应尽量使配制的对照品溶液的浓度与样品中组分的浓度相近。 外标法是色谱分析中的一种定量方法,它不是把标准物质加入到被测样品中,而是在与被测样品相同的色谱条件下单独测定,把得到的色谱峰面积与被测组分的色谱峰面积进行比较求得被测组分的含量。外标物与被测组分同为一种物质但要求它有一定的纯度,分析时外标物的浓度应与被测物浓度相接近,以利于定量分析的准确性。 [url=http://www.gdkjfw.com/][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url][/url]简单的说:内标法就是用在样品中定量加入要分析的物质,通过测得的实际样品量和加入样品量的比值来定量所要分析的样品含量。 内标法主要优点是简单,快速。 缺点是没有标准曲线法定量精确。[align=center][/align]
气相色谱法是近代仪器分析方法之一。它在分析化学中占有一定的地位。但是气相色谱法决不是万能的,在很多场合下,它必须与其他仪器配合,才能解决问题。因此,要根据具体分析对象,选择合理的分析方法。 1.与化学分析法比较 化学分析是按物质的特殊化学反应进行分析的。在这一方面前人已积姨了丰富的经验,大部分方法亦属于经典方法。其特点是所用仪器简单、价廉、操作也不复杂,且可进行同族、同系物的总含量测定(如滴定、氧化、还原等方法),对于单个组份的测定,准确可靠,故可作气相色谱法的对照、旁证方法。其缺点是不能测定化学性质迟钝或性质极为相近的复杂物质。气相色谱法分析这类物质却轻而易举。但色谱定量时要做校正因子、校正曲线,即使只分析一个样品也要这样做,故建立方法费时。色谱法难以分析腐蚀性或反应性较强的物质,如HF, OE、过氧化物等,而化学法分析则甚为简便。另外,在处理一些特殊样品的定性、定量工作中,亦需与化学法结合起来才能解决。如经基的脂化、.经基的硅醚化、二次加工、油品的酸碱处理等。所以需要求购仪器仪表,这样有实际的比照才会做出明显的区别。 2.与光谱、质谱法比较 气相色谱法的最大优点是易分离。分析多组份混合物,光谱(红外,紫外光谱)、质谱法就不及色谱法。而且一般来说色谱法的灵敏度与质谱接近,比光谱要高,造价却比光谱、质谱仪都低。色谱法的缺点主要是难以对未知物分析定性,如果没有已知的纯样品或已知纯样品的色谱图,就很难判断某一色谱峰究竟代表何物。而质谱则既能分析多组份混合物,且可测定出未知物的分子沮。用光谱法可以测出分子中含有那些官能团。这些都是气相色谱法所不及的。所以把色谱与质谱、光谱结合起来联用,就可以解决未知物的分析问题,发挥更大的作用,成为目前解决复杂混合物强有力的先进手段之一。这种结合包括收集色谱分离后的单组份或窄馏份,用光谱、质谱定性。色谱一质谱联用,色谱一光谱联用等。国内利用毛细管色谱一质谱联用仪成功地解决了一些油品的组份分析。不论从速度或效果看,都是十分理想的。比如最好的鉴别仪器是在线微波水分仪等等。 3.与精密分馏比较 色谱柱的效能和精馏塔一样,也是用理论板数来度量。但获得某一纯度分离所需要的板数,色谱法比精馏法要高得多。例如:分离同一有具体名称的样品,精馏塔需要100块塔板,色谱柱则需要10000块塔板。这是因为色谱柱中每一时刻都只有某一小部分柱在起分离作用,而精馏中却是在全部时间里全部塔板同时起分离作用。但提高色谱柱理论板数是较容易实现的,因此,用大型制备色谱可以制出纯度高达99.99%的纯物质,比精馏产品纯度高得多,所需时间也较短,但处理量小是其不足之处。 4.与经典的测定物化常数比较经典法测定物化常数,通常手续麻烦,时间较长,且需用纯物质。气相色谱法的特点是设备简单,操作方便,可以同时测定两种或多种物质相差微小的物化常数,如分配系数、活度系数、溶解热、自由能、自由摘等,而且不必分离杂质,一次可测出多种数据。但色谱法的缺点是要作一些简化假设(如载体不起作用是惰性的等),数学处理较复杂,数据精度也较差。
最近做一个课题,关于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]技术在农药残留分析的应用及发展!本人才疏学浅跪请各位大侠能拔刀相助,救在下一马!十万火急!!![em53]
色谱分析法又称层析分析法,是一种分离测定多组分混合物的极其有效的分析方法。其原理是:不同物质在相对运动的两相中具有不同的分配系数,当这些物质随流动相移动时,就在两相之间进行反复多次分配,使原来分配系数只有微小差异的各组分得到很好地分离,依次送入检测器测定,达到分离、分析各组分的目的。色谱法的分类方法很多,常按两相所处的状态,可分为气相色谱(用气体作为流动相)和液相色谱(用液体作为流动相)。液相色谱又可分为柱层析、纸层析、薄层层析和高效液相色谱分析。气相色谱法是使用气相色谱仪来实现对多组分混合物分离和分析的。载气由高压钢瓶供给,经减压、干燥、净化和测量流量后进入气化室,携带由气化室进样口注入并迅速气化为蒸气的试样进入色谱柱(内装固定相),经分离后的各组分依次进入检测器,将浓度或质量信号转换成电信号,经阻抗转化和放大,送人记录仪记录色谱峰如果分离完全,每个色谱峰代表一种组分。根据色谱峰出峰时间可进行定性分析;根据色谱峰高或峰面积可进行定量分析。
我用SP 6800A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](FID)分析样品,其他条件未改变,改变分流比会得到不同的分析结果(面积归一法),而且相差较大 ,都在检测限内,请问是什么原因?
讨论: 工业乙炔中痕量有机气体、无机气体分析 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法成份O2N2 CO2 CO CH4C2H6C2H4C3H8 丙酮C2H2 检测限ppm250.50.50.51112090%--99.99% 六通阀进样,反吹,氢气作载气,1m5A分子筛,分离O2、N2 ,TCD检测;2m Porapark Q ,N2 作载气,分离CO2、CO、CH4 ,甲烷转化炉,FID检测;十通阀进样,Porapark Q预柱,N2 作载气,中心切割,POLT Al2O3 分离,FID检测。上述只是个人意见,不足之处还望批评指正!!
鬼峰或交叉污染系统的污染主要是由鬼峰或交叉污染造成的。如果鬼峰的峰宽与样品的峰类似(具有类似的保留时间),则污染物很可能是与样品同时进入色谱柱的。进样器中可能存在额外的化合物(即污染物)或样品本身存在这些化合物。溶剂、样品瓶、瓶盖和注射器中的杂质只是某些可能的污染源。进样样品和溶剂空白有助于找到可能的污染物源。如果鬼峰的峰宽比样品峰宽很多,则污染物极可能在进样样品时已存在于色谱柱中了。这些化合物在上一次GC进样结束时已存在于色变柱中了。在下一次进样时这些化合物会流出,因而峰很宽。有时,一些鬼峰是由多次进样累积而成的,因此流出时呈现圆丘峰或圆包峰。这样的鬼峰常常随基线的漂移或偏移而出现。提高升温程序中的最高温度或延长升温时间是减少或消除鬼峰问题的方法之一。另外,在每次进样后或序列分析后进行短暂的烘烤,也可以从色谱柱中去除保留性较强的化合物,从面避免导致出现问题。浓缩测试如果怀疑进样器或载气存在被污染的问题(例如有鬼峰或基线不稳定),可使用这一方法进行测试。将GC在40-50℃下运行8小时或更长时间在正常的温度条件和仪器设定条件下进行空白分析(即启动GC但不进样)采集这一空白试验的色谱图在第一次试验完成后,立即重复进行一次空白试验。必须在5分钟内开始进行第二次空白试验。采集第二次空白试验的色谱图,并将其与第一次的色谱图进行比较如果第二次试验的色谱图明显有大量的峰出现并且基线也不稳定,则表明载气路或载气已被污染。如果第二次试验的色谱图中只有很少的峰出现并且基线也没有明显的漂移,则表明进入的载气或载气气路比较干净。
当[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]衬管或者进样口被污染时,常常会造成样品色谱特征的变化, 如导致峰不对称,谱图变形等。为保证仪器的分离效果以及分析结果的精密度、准确性,需要经常保养衬管和进样管。 衬管的结构要求和尺寸影响进样效果,进一步影响分析结果,具体表现在以下几个方面: 1.衬管的内被污染,或有进样隔垫碎屑,易导致分析结果严重性差(如保留时间、峰面积等的重现性)或出现鬼峰; 2.衬管内的石英玻璃毛等装填物填装不当,或装填物、衬管内壁有活性点,容易导致实验结果的重复性差; 3.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]长时间运行后,进样口经常残留有凝固的样品。当进样口有样品残留时, a如果凝固样品残留在进行隔垫上面或下面,容易导致进样器进样时针尖不好插入。 b如凝固样品残留在分流管路部分内部,会导致管路内径变细,甚至堵塞。 c当进样口温度比通常工作温度高很多时,之前凝固样品可能会出现鬼峰,影响分析结果的重现性。在日常使用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]时,衬管的定期保养非常重要。
[color=#444444]本人想用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析H2,乙烯,乙烷,乙炔,有没有单独使用一种检测器就可以分析全部成分的方法,色谱柱和检测器等分析条件介绍越详细越好,谢谢啦!!![/color]
选择[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的实验条件主要包括:色谱柱的选择、柱温的选择和载气的选择。(1)色谱柱的选择主要是选择固定相和柱长。固定相选择需注意极性及最高使用温度。气一液色谱法还要注意载体的选择。高沸点样品用比表面小的载体、低固定液配比(1%-3%),以防保留时间过长,峰扩张严重。低沸点样品宜用高固定液配比(5%-25%),从而增大分配系数,以达到良好分离。难分离样品可用毛细管柱。柱长加长能增加塔板数,使分离度提高。但柱长过长,峰变宽,柱阻也增加,并不利于分离。在不改变塔板高度(H)的条件下,分离度与柱长有如下关系。(2)柱温的选择选择的基本原则是:在使最难分离的组分有符合要求的分离度的前提下,尽可能采用较低柱温。低柱温可增大分配系数,增加选择性,减少固定液流失,延长柱寿命及降低检测本底。但柱温降低,液相传质阻抗增加,而使峰扩张,柱温太低则拖尾,故以不拖尾为度。可根据样品沸点来选择柱温。分离高沸点样品(300-400℃),柱温可比沸点低100-150℃。分离沸点300℃的样品,柱温可以在比平均沸点低50℃:至平均沸点的温度范围内。对于宽沸程样品(混合物中高沸点组分与低沸点组分的沸点之差称为沸程),选择一个恒柱温经常不能兼顾两头,需采取程序升温的方法。程序升温改善了复杂成分样品的分离效果,使各成分都能在较佳的温度下分离。程序升温还能缩短分析周期,改善峰形,提高环境监测中检测灵敏度。(3)载气的选择载气的选择从三方面考虑:对峰扩张、柱压降及环境监测中检测器灵敏度的影响。载气采用低线速时,宜用氮气为载气,高线速时宜用氢气(黏度小)。色谱柱较长时,在柱内产生较大的压力降,此时采用黏度低的氢气较合适。H2最佳线速度为10-12cm/s;N2为7-10cm/s。通常载气流速可在20-80mL/min内,通过实验确定最佳流速,以获得高柱效,但为缩短分析时间,载气流速常高于最佳流速。
1900mV。mL/mL。 4。2正确度 表2列出了实验所测得的正确度的具体数据。 4。3最低检测浓度 在实验条件下实测最低检测浓度见表3 5结论 (1)硫化物分析可采用气固和气液色谱法,但在工艺控制分析中采用气固色谱法具有无固定相流失、柱寿命长等特点,同时,也不会因柱室超温而损坏柱子,可用于高灵敏度检测器。实验证实GDX-301是分析硫化物的理想柱子; (2)由于硫化氢、二氧化硫、二硫化碳和硫氧碳都是极性物质,在分析过程中轻易发生吸附现象而影响分析结果。因此,应尽量缩短采样时间,确保分析结果的正确性; 3在分析低含量氧时,应采取有效的密封措施,防止空气中氧的渗透。 该方法是为硫磺回收过程气的分析而开发的,方法快速、正确、灵敏,分析误差为±1。4%,试验证实满足硫回收过程气的工艺分析。
[color=#444444]采用内标法[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析物质,如何进行校正?例如[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析甲酸,甲酸的响应信号不好,将甲酸和醇反应成甲酸乙酯,再通过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析甲酸乙酯的含量,反推出甲酸的含量。绘制了甲酸乙酯的标准曲线,如今通过反推甲酸有1.8 g,但甲酸的实际量有2.0 g,这样的话如何校正?有没有类似的文献,急求!![/color]
在实际工作中,当我们拿到一个样品,我们该怎样如何定性和定量,建立一套完整的分析方法是关键,下面介绍一些常规的步骤:1、样品的来源和预处理方法GC能直接分析的样品必须是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐),可能会损坏色谱柱的组分。这样,我们在接到一个未知样品时,就必须了解的来源,从而估计样品可能含有的组分,以及样品的沸点范围。如能确认样品可直接分析。如果样品中有不能用GC直接分析的组分,或样品浓度太低,就必须进行必要的预处理,包括采用一些预分离手段,如各种萃取技术、浓缩和稀释方法、提纯方法等。2、确定仪器配置所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。3、确定初始操作条件当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。这时要确定初始分离条件,主要包括进样量、进样口温度、检测器温度、色谱柱温度和载气流速。进样量要根据样品浓度、色谱柱容量和检测器灵敏度来确定。样品浓度不超过mg/mL时填充柱的进样量通常为1-5uL,而对于毛细管柱,若分流比为50:1时,进样量一般不超过2uL。进样口温度主要由样品的沸点范围决定,还要考虑色谱柱的使用温度。原则上讲,进样口温度高一些有利,一般要接近样品中沸点最高的组分的沸点,但要低于易分解温度。4、分离条件优化分离条件优化目的就是要在最短的分析时间内达到符合要求的分离结果。在改变柱温和载气流速也达不到基线分离的目的时,就应更换更长的色谱柱,甚至更换不同固定相的色谱柱,因为在GC中,色谱柱是分离成败的关键。5、定性鉴定所谓定性鉴定就是确定色谱峰的归属。对于简单的样品,可通过标准物质对照来定性。就是在相同的色谱条件下,分别注射标准样品和实际样品,根据保留值即可确定色谱图上哪个峰是要分析的组分。定性时必须注意,在同一色谱柱上,不同化合物可能有相同的保留值,所以,对未知样品的定性仅仅用一个保留数据是不够的,双柱或多柱保留指数定性是GC中较为可靠的方法,因为不同的化合物在不同的色谱柱上具有相同保留值的几率要小得多。6、定量分析要确定用什么定量方法来测定待测组分的含量。常用的色谱定量方法不外乎峰面积(峰高)百分比法、归一化法、内标法、外标法和标准加入法(又叫叠加法)。峰面积(峰高)百分比法最简单,但最不准确。只有样品由同系物组成、或者只是为了粗略地定量时该法才是可选择的。相比而言,内标法的定量精度最高,因为它是用相对于标准物(叫内标物)的响应值来定量的,而内标物要分别加到标准样品和未知样品中,这样就可抵消由于操作条件(包括进样量)的波动带来的误差。至于标准加入法,是在未知样品中定量加入待测物的标准品,然后根据峰面积(或峰高)的增加量来进行定量计算。其样品制备过程与内标法类似但计算原理则完全是来自外标法。标准加入法定量精度应该介于内标法和外标法之间。7、方法的验证所谓的方法验证,就是要证明所开发方法的实用性和可靠性。实用性一般指所用仪器配置是否全部可作为商品购得,样品处理方法是否简单易操作,分析时间是否合理,分析成本是否可被同行接受等。可靠性则包括定量的线性范围、检测限、方法回收率、重复性、重现性和准确度等。本文摘自《气相色谱方法及应用》
[color=#444444]最近看了一篇文献,是液体a和液体b反应得到液体c的问题,文献上说用“取反应液用 9790 型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]分析(OV-101[/color][color=#444444]毛细管柱,氢火焰离子化检测器)测定,用峰面积归一法确定产率。“[/color][color=#444444]请问,是怎么计算产率的,能不能提供点知识?[/color]
用气相色谱法做痕量分析是一门综合的实验技术。可以把痕量色谱分析过程归纳以下四个阶段: ①样品采集; ②样品制备(予处理); ③色谱分析; ④数据处理与结果表达。 如果样品采集和前处理比较成功,在色谱分析和数据处理时,即使选用的色谱仪所配用的检测器灵敏度不高,分析柱分离效率较低,数据处理装置性能、功能一般,也能获得比较理想的实验结果。反过来说,若所选购的仪器和数据处理装置、配置较高又选择了一根高效色谱柱,那么可大大降低样品的予处理过程。在目前的痕量分析中,耗时、费力和效率低的样品采集与处理仍是整个色谱分析中的瓶颈。样品采集和处理时间有时要占了整个分析时间的三分之二。 应当指出,无论是何种最先进的色谱仪和设备,真正高性能色谱柱,最完善的数据处理装置,都不能从一个采集处理不适当样品得到满意的分析结果。因此,在选购仪器(含数据处理装置)类型和性能时,要考虑如何充分发挥所选仪器的综合分析能力,以便简化样品的予处理过程或根本不需要样品的予处理。
色谱分析定量方法为归一法,分析结果如何以算术平均值报出?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法及其在水质分析中的应用(1) [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法是50年代后迅速发展起来的一种对复杂混合物中各种组分的分离和分析技术。由于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法具有高效能、高选择性、高灵敏度、分析速度快、样品用量小、设备简单、投资小、应用范围广等优点。因此[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]自1954年商品化以来,发展速度之快是少有的。在不断发展、丰富和提高的过程中又与质谱、傅利叶变换红外光谱、计算机联用,更加扩展、完善和提高了对复杂多组分混合物进行定性、定量分析的用途。最近几年,各种新型气相色谱仪,鉴定器,高效、耐高温色谱柱的出现;超临界流体色谱的研究和迅速发展,更加拓宽了应用范围。在近代分析工作中,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的地位如同经典分析化学中的天平、生物研究中的显微镜一样重要。目前已广泛用于石油、化工、医药、卫生、有机合成、生物化学、食品、农药及环境监测等领域。 (一)[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]发展史 50年代初期,随着新兴石油工业的出现以及医药、生物化学的发展,促进了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的产生和发展,借助于先进的电子技术又使[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]日益完善。所以[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]问世以后很快威为分析领域中极为重要的分析手段。 1906年俄国植物学家’Fswett利用液固色谱法研究了植物的绿叶成分。1941年Martin和Synge提出了液液色谱法,预见了气液色谱。1952年James和Martin提出了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法。1954年瑞依把热导检测器用于气液色谱法。1957年Golay提出了毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,也叫作开管柱[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法。1958年McWilliam报导了氢火焰离子化检测器(FID)。1979年Dandeneau等拉制出石英弹性毛细管柱,从而促进了毛细管色谱法的大发展。进入80年代以后,微处理计算机在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]之应用、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]一质谱联用、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]一傅利叶变换红外光谱联用、超临界流体色谱的迅速发展,都大大地扩展了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]科学的内容和应用范围。(二)[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的原理 当载气把被分析的气态混合物带入装有固定相的色谱柱时,由于各组分的分子与固定相分子间发生吸附、脱附或溶解、离子交换等物理化学过程,使各组分的分子在载气和固定相的两相间分配系数不一样,经反复多次分配,不同组分在色谱柱上移动速度不一样,经过龟谱柱后,各组分按先后次序流出色谱柱,使各组分得到完全分离。(三)[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的特点 (1)分离效能高。一般填充柱有几千块理论塔板,而毛细管柱理论塔板达103-106,因而可以分析沸点相近的组分和十分复杂的混合物。例如60m长的毛细管柱可将汽油含10个碳以内的组分分离出240个色谱峰。 (2)选择性高。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法可以分离化学结构极为相近的化合物。例如多环芳烃的异构体、二甲苯的三种异构体等,用其它方法分离相当困难,但[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法则比较容易。 (3)高灵敏度。与[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]配用的高灵敏检测器对待测物的最低检测质量可达0.1-10pg或更低。因而适合于微量有机物分析,例如采用电子捕获检测器,可测0.1.pgγ/-666。 (4)分析速度快。一次色谱分析短者几秒钟,长者几分钟到几十分钟完成。特别是目前[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]可由微处理机控制并配数字处理系统,速度就更快。 (5)样品用量少。由于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]灵敏度高,需要的样品极少,一般进样几个微升即能完成全分析。 (6)分离和检测能一次完成,这也是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的特点。其它的分析方法,如紫外、红外、质谱等均需纯品,对混合物则需分步进行分离和检测。但是,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的应用有其局限性。即只能测定单一物质的量,不能测定某些同类物的总量;在进行定性和定量分析时,需要被测物的标准品为对照,而标准品往往不易获得,这给定性鉴定带来困难。(四)[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]常用的名词、术语及定义 (1)色谱图 样品各组分经色谱柱分离后进人检测器,它们在检测器中引起响应并作为分离时间的函数被记录下来,这样一系列表示组分性质、含量信号一时间曲线就是色谱图。对于微分型的检测器,信号近似于正态分布曲线,色谱峰面积正比于组分质量。色谱图是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法定性、定量的依据,也是衡量仪器好坏的依据。 (2)基线 在操作条件下,只有纯载气经过检测器时,记录仪所记录的检测器输出的信号一时间曲线为基线。理论上是一条直线,但在高灵敏度量程时,基线常有一定的噪声和漂移。噪声:基线在短暂时间的波动,以波动的峰值表示。噪声大往往和基流高联系在一起。漂移:基线在一段较长时间(如几十分钟)内缓慢改变。噪声是叠加在漂移上同时表现出来。 (3)色谱峰 在操作条件下,当载气带着样品组分经过检测器时,检测器输出的信号随着时间变化的曲线为色谱峰。理想的色谱峰为正态分布函数,所表示的峰形是对称的。 峰高:色谱峰的最高点到峰底(峰下面基线的延伸部分)的垂直距离,一般常用h表示。 半峰高宽度:峰高1/2处的宽度,常用单位为cm。 峰面积:指色谱流出曲线与基线之间所包含的面积
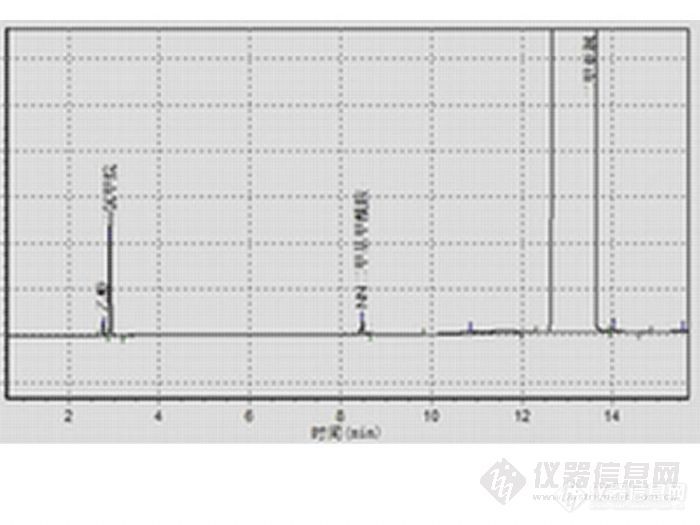
顶空毛细管柱气相色谱法分析测定药品中残留溶剂http://ng1.17img.cn/bbsfiles/images/2011/05/201105211013_295254_2242538_3.jpg 摘要 目前国家对药品中残留溶剂的检测还没有一个统一的国家标准,在关于有机溶剂残留量的指导原则中将二氯甲烷、DMF列为第二类必须控制的毒性试剂,将乙醇、丙酮列为第三类低毒性的溶剂。建立一种快速测定药品中残留溶剂的分析方法很有必要。为此南京科捷采用DK-300A顶空进样器取样,气相色谱毛细管柱分离,分析测定药品中常见的残留溶剂(乙醇、二氯甲烷、NN二甲基甲酰胺、二甲亚枫)。结合相关指标,本实验将二氯甲烷的限量规定为 600ppm,DMF的限量规定为 880ppm,乙醇及丙酮的限量规定为 5000ppm。实验结果表明:顶空毛细管柱气相色谱法快速、简便、准确是测定药品中残留溶剂较为理想的方法。关键词 乙醇 二氯甲烷 NN二甲基甲酰胺 顶空进样 毛细管气相色谱 药品 有机溶剂残留1.药品中乙醇、二氯甲烷、NN二甲基甲酰胺色谱图峰序1. 乙醇2.二氯甲烷3.NN二甲基甲酰胺4.二甲亚枫(溶剂)2.本方法应用范围在合成原料药,辅料或制剂生产的过程中使用或产生的挥发性有机化学物质,它们在实际的生产中未能被完全地清除。本方法可应用在药品中残留溶剂的检测中。近年来,药品中残留有机溶剂的毒性和致癌作用日益引起各方面的重视。药品中残留有机溶剂于1997年被美国FDA列为药品监控项目。我国药品中残留有机溶剂检测也越来越受到有关方面的重视。本文初步研究探索采用顶空毛细管柱气相色谱法分析药品中残留挥发性有机溶剂。顶空气相色谱法只将挥发和半挥发的组份引入柱子,可避免非挥发性的物质对系统的污染,样品前处理简便,分析效率高。结果表明,本方法快速、准确、重现性好。3.仪器及试剂配置色谱仪器配置色谱柱及试剂GC5890(FID检测器)毛细管专用柱30*0.32.*0.5乙醇、二氯甲烷各一瓶顶空进样器:DK-300ANN二甲基甲酰胺1瓶N2000色谱工作站(电脑自备1台)二甲亚枫1瓶氢氮氧一体发生器或钢瓶气各一瓶顶空压盖机1台顶空瓶20ml (带塞) 50只







 我要推广仪器
我要推广仪器
 下载APP
下载APP